ALZHEIMER'S DISEASE
Alzheimer's Disease (AD) is a specific neurodegenerative disease and is the most common cause of dementia in old people. Clinically, it is characterized by loss of memory, inability to learn new things, loss of language function, a deranged perception of space, inability to do calculations, indifference, depression, delusions, and other manifestations. These deficits affect patients' social functioning and make it difficult or impossible for them to carry on with daily living. AD is inexorably progressive and fatal within 5 to 10 years. AD patients usually die of complications of chronic illness. AD is the fourth to fifth most common cause of death in the United States. Sometimes AD involves people in their 40s and 50s, but is mainly a disease of old age. Based on clinical evaluations, 13% of persons over 65 years and 45% of those over 85 have AD.
Mild cognitive impairment (MCI) is a clinical state characterized by memory and other cognitive impairments that do not affect social functioning or daily living. Some MCI patients progress to AD but others remain stable or revert to normal, indicating that MCI has diverse causes.
PATHOGENESIS OF AD
AD is driven by two processes: extracellular deposition of beta amyloid-Aβ and intracellular accumulation of tau protein. Both these compounds are insoluble. Aβ is the main component of senile plaques and tau is the component of neurofibrillary tangles. Aβ deposition is specific for AD and is thought to be primary. Tau accumulation is also seen in other degenerative diseases and is thought to be secondary.
BETA AMYLOID. Aβ is a 36 to 43 amino acid peptide, which is part of a larger protein, the Amyloid Precursor Protein (APP). APP is a transmembrane protein, made by neurons and other brain cells. It is also found in extraneural tissues and is especially abundant in platelets. Its function is unknown. The Aβ amyloid residue includes part of the transmembrane domain of APP and is derived from cleavage of APP by the enzymes β-and γ-secretase. Aβ monomers and oligomers are further degraded by other enzymes. Defective clearance of Aβ from aberrant cleavage of APP and other mechanisms (see genetics, further on) results in its accumulation. Aβ monomers polymerize initially into soluble oligomers and then into larger insoluble fragments such as Aβ42, which precipitate as amyloid fibrils.
Aβ is toxic to neurons. In brain slice preparations, it causes loss of long term potentiation, damages synapses, and kills neurons. Moreover, it shows selective neurotoxicity for the hippocampus and entorhinal cortex (areas that are severely affected in AD) while sparing cerebellar neurons. This damage is mediated by free radicals, which are generated when soluble Aβ is complexed with Zn2+, Cu2+, and Fe3+. There is a high correlation between the amount of soluble Aβ and the severity of the neurological dysfunction in AD. In transgenic AD models, severe neurological deficits occur in absence of amyloid deposits in tissue.
The beta amyloid hypothesis is the basis of a novel prevention and treatment method for AD that was reported recently in transgenic mice that overexpress a mutant APP and develop AD neuropathology. Active immunization of young animals with Aβ and passive immunization with Aβ antibodies prevented the development of AD; immunization of older animals reduced the extent and severity of AD pathology. Based on these findings, a human vaccine consisting of synthetic Aβ was developed. Phase I trials went uneventfully but phase II studies were terminated because some patients developed an autoimmune meningoencepalitis.
TAU. Neurofibrillary degeneration is characterized by the deposition in the neuronal body and processes of insoluble polymers of over-phosphorylated microtubule associated protein tau. Tau aggregates as pairs of filaments that are twisted around one another (paired helical filaments). These deposits interfere with cellular functions by displacing organelles. By distorting the spacing of microtubules, they impair the axonal transport thus affecting the nutrition of axon terminals and dendrites. No mutations of the tau gene occur in AD. Abnormal tau first appears in the entorhinal cortex, then in the hippocampus, and at later stages in association cortex. Recent observations in transgenic mice suggest that the spread of the pathology to anatomically linked areas occurs by passage of abnormal tau across synapses.
PATHOLOGY OF AD
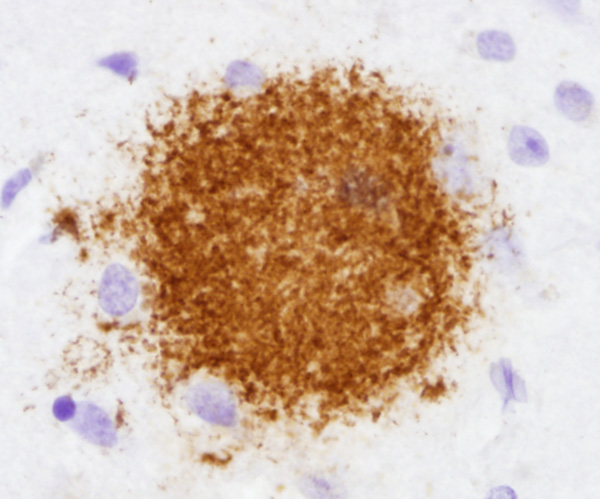 Diffuse plaque |
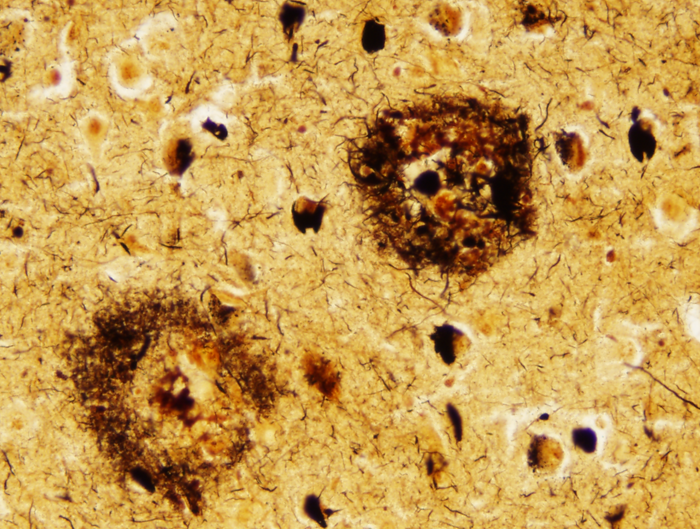 Senile plaques. Bielschowski silver stain. |
 AD. Gliosis. |
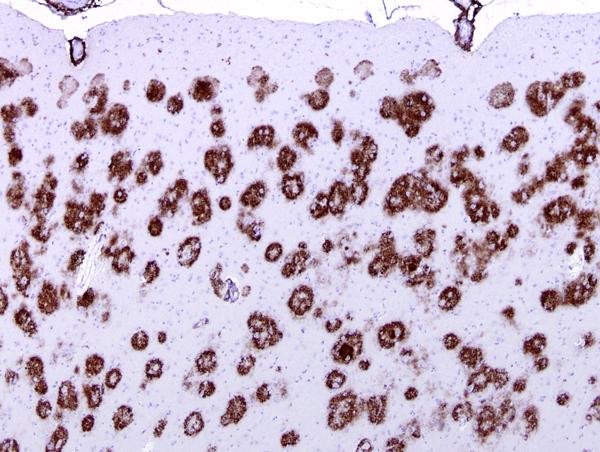 Amyloid in plaques and vessels in the cerebral cortex |
There are two main lesions in AD, senile plaques (SPs) (also called Alzheimer's plaques) which contain Aβ, and neurofibrillary tangles (NFTs), which contain over-phosphorylated tau. There are 2 kinds of SPs: diffuse Aβ plaques (AβPs) and neuritic plaques (NPs). AβPs are spherical exracellular Aβ deposits. NPs are AβPs containing degenerating neuronal processes with tau paired helical filaments. Aβ in NPs frequently forms a central core or small chunks, has a fibrillary fine structure and is Congo Red positive and birefringent, similar to other amyloids. NPs contain also reactive astrocytes and microglia, indicating an inflammatory reaction. Diffuse plaques do not disrupt the neuropil. They are seen, sometimes in large numbers, in old, non-demented persons and are not associated with dementia. Aβ deposition in AD appears first in the neocortex and as the disease progresses, it spreads to allocortex, deep nuclei, and, in severe cases, it may extend to the brainstem and cerebellum.
 Neurofibrillary tangle and neuritic plaque |
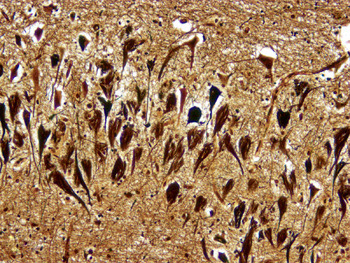 Neurofibrillary tangles |
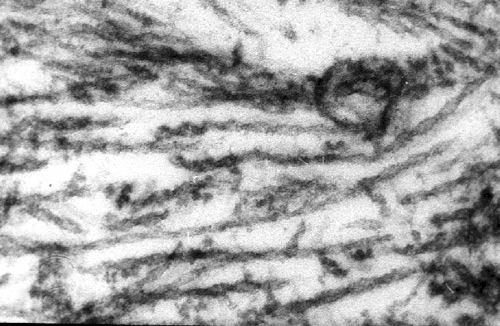 Paired helical filaments |
NFTs are deposits of tau filaments in the neuronal body. Similar deposits are present in the dystrophic processes of NPs and in dendrites (neuropil threads-NTs). In advanced AD, the hippocampus often contains extracellular NFTs embedded in the neuropil, like fossilized skeletons of neurons (ghost tangles). NFTs appear first in the transentorhinal region of the temporal lobe, lateral to the hippocampus, and then spread to the entorhinal cortex, hippocampus, and association neocortex.The mechanism of accumulation of tau in NFTs is unclear. However, NFTs develop independently of Aβ, and cognitive decline correlates better with NFT load rather than with the number of SPs. On the other hand, genetic and environmental risks for AD (see further on) have a stronger association with amyloid.
SPs are only found in AD. NFTS are found in many neurodegenerative diseases besides AD, including the frontotemporal dementias, chronic traumatic encephalopathy, myotonic dystrophy, and prion diseases. This indicates that NFTs can cause neurodegeneration indepenently of Aβ deposition. Most cases of AD show a combination of SPs and NFTs, but some cases have a predominance of one or the other. In very severe cases, SPs and NFTs appear also in deep nuclei and in the brain stem. The prevailing opinion is that the primary lesion in AD is Aβ deposition and NFTs are secondary. There are no such lesions in the white matter.
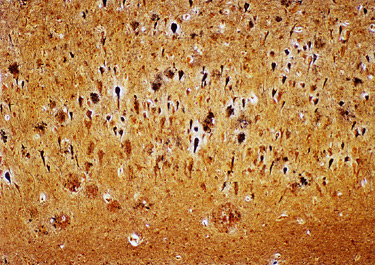 Large numbers of plaques and tangles |
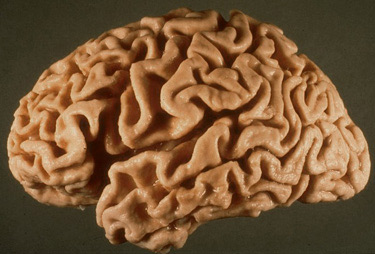 Cortical atrophy |
 Cortical atrophy and hydrocephalus ex vacuo |
Each SP represents a focus of damage of the neuropil that includes axon terminals and dendrites of several neurons and probably thousands of synapses. NFTs also cause neuronal dysfunction and loss. Thus, the result of these lesions is severe disconnection. The distribution of the lesions correlates with the clinical picture. Damage of the hippocampus explains the impairment of memory, and involvement of association cortex correlates with the loss of higher intellectual functions. The loss of neurons and synapses that are associated with SPs and NFTs causes cortical atrophy, and dilatation of the lateral ventricles due to loss of brain tissue (hydrocephalus ex vacuo).
The lesions of AD can be best appreciated in sections of brain stained with the Bielschowski silver stain, the fluorescent stain thioflavin S, and immunostains for Aβ. NFTs and neuropil threads fluoresce and stain black with the Bielschowski stain. Aβ immunostains highlight the amyloid cores of SPs and reveal also diffuse amyloid deposits without disruption of the neuropil (diffuse plaques).
Other frequent neuropathological findings in AD are granulovacuolar degeneration (GVD) and Hirano bodies (HB), seen mainly in pyramidal neurons of the hippocampus. GVD consists of intracytoplasmic vacuoles containing basophilic granules. HBs are eosinophilic cytoplasmic neuronal inclusions of actin and actin-binding proteins. The pathogenesis of these lesions and their contribution to the neurodegeneration of AD is unclear. I addition, many AD patients have also Parkinson’s disease, TDP-43 deposits, and other tauopathies.
NEUROPATHOLOGICAL STAGING OF AD
|
The Consortium to Establish a Registry for Alzheimer's disease (CERAD) developed a neuritic plaque scoring system, which ranks the density of neuritic plaques identified by Bielschowski silver stains in the neocortex. The Braak and Braak staging scheme focuses on NFTs. Stages I and II, in this system, correspond to transentorhinal involvement; stages III and IV to limbic; and V and VI to isocortical involvement. NFTs in the hippocampus and entorhinal cortex correlate with memory impairment; neocortical NFTs correlate with cognitive decline. The Thal system describes 5 phases of beta amyloid deposition. In phase 1, only the neocortex is affected. Progressively, amyloid spreads to allocortex, subcortical nuclei, brainstem, and in phase 5 it involves also the cerebellum. The 2012 National Institute on Aging-Alzheimer’s Association (NIA-AA) criteria for neuropathological diagnosis of AD take into account Aβ plaque score (Thal phase), NFT stage (Braak stage), and neuritic plaque score (CERAD score). |
ETIOLOGY OF AD-GENETICS
The gene
for APP is on chromosome 21. Trisomy 21 (Down syndrome)
provides a clear mechanism for Aβ deposition.
Persons with this condition produce one and a half
times as much APP as normal people do and develop AD
at a young age, some of them in their 20s. Most AD,
however, is not due to overproduction of APP, but to
some other mechanism, either an abnormality of the
APP molecule that renders it more amyloidogenic, or
a defect of processing of normal APP. This appears
to be the case in infrequent genetic forms of AD. In
these patients, autosomal dominant AD develops before
age 65 (presenile dementia) due to mutations of the
APP gene and of the presenillin1
and 2 genes (PSEN1 and PSEN2) on chromosomes 14 and 1 respectively. The
presenillins are catalytic components of γ-secretase.
Setting aside
Down syndrome and autosomal dominant AD, the vast
majority (95%) of AD cases appear in old age and
do not follow Mendelian inheritance. These cases are
due to an interaction of genetic and other
intrinsic and environmental factors. Genetics accounts
for 70% of the risk, and the most important genetic
risk factor is the Apolipoprotein E (ApoE) genotype.
ApoE is a protein that carries cholesterol in and
out of cells. It occurs in three isoforms: ApoE2,
ApoE3, and ApoE4. The gene for ApoE is on chromosome
19. One copy is inherited from each parent. The ApoE4
allele confers a high risk for AD, the rare ApoE 2
confers a low risk, and the most common ApoE3 an intermediate
risk. About 63% of Europeans
are ε3/ε3 and 19% are ε3/ε4. Persons who
are homozygous for the ApoE4 allele develop AD at
a mean age 70. Persons with other ApoE genotypes develop
the disease later or not at all. However, having an
ApoE4 allele does not mean that one will invariably
develop AD. Furthermore, the role of ApoE4 in non-Europeans
is not as well established. The ApoE4 allele is also
a risk factor for hypercholesterolemia. High cholesterol
levels during mid-life increase the risk of AD and
lipid-lowering drugs lower this risk. ApoE4 has been
detected in NFTs and in Aβ. These
findings suggest that ApoE lipoproteins participate
in some way in the processing of APP, but the mechanism by which ApoE4 induces a high risk for AD is unclear.
ETIOLOGY OF AD-OTHER CONTRIBUTING FACTORS
The role of the environment, diet, and general state of health in AD is being actively explored. Chronic cellular damage from free radicals, excitotoxicity, nonenzymatic glycation of proteins, and other factors contributes to the loss of neurons and synapses that is associated with old age and aggravates the pathology of AD.
Neuroinflammation. Other than Aβ and tau, neuroinflammation is the most important factor involved in the pathogenesis of AD. There are multiple findings underlining the role of neuroinflammation. Acute-phase proteins are elevated in serum and deposited in SPs; microglial cells accumulate around SPs; and complement components are present in SPs. APP is an acute phase protein which is released in brain tissue following trauma and other insults. The effects of neuroinflammation are mediated by activated microglial cells which are a source of cytokines and a potent generator of free radicals. Advanced molecular studies have revealed multiple aberrations in the microglial immune response in AD. These aberrations are triggered by Aβ and tau but, in turn, cause brain damage and accelerate Aβ and tau deposition.
Free radicals: Oxidative stress, compounding with advancing age, causes mitochondrial DNA mutations, mitochondrial dysfunction and more oxidative stress. This process is accelerated in AD by the action of Aβ (a mitochondrial poison and free radical generator) and activated microglia, also a source of free radicals.
Diabetes: Type 2 diabetes is a risk factor for AD. AD patients have low levels of insulin and insulin resistance in the brain. These changes impair energy metabolism in neurons and adversely effect signaling pathways dependent on insulin and its receptors. Furthermore, nonenzymatic glycation of proteins produces neurotoxic derivatives that aggravate oxidative damage.
Traumatic brain injury: Dementia and parkinsonism develop sometimes in boxers, football players and other individuals who have had repeated cerebral concussions. This entity has been previously called dementia pugilistica (or the punch drunk syndrome) and was recently renamed chronic traumatic encephalopathy (CTE). The brain in CTE shows mainly NFTs. Diffuse and less frequently neuritic plaques are seen inconstantly.
Homocysteine. Increased levels of homocysteine (also a risk factor for stroke) and decreased dietary folate potentiate these neurotoxic effects. Homocysteine increases with advancing age and is elevated in persons with polymorphisms of 5,10-methylenetetrahydrofolate reductase (MTHFR), an important enzyme involved in folate metabolism. Such polymorphisms are very common. Elevated homocysteine and decreased folate are associated with increased free radicals, cytosolic calcium, glutamate excitotoxicity, apoptosis, and decreased levels of ATP.
OLD AGE AND AD
The fact that many people in their 80s and 90s are mentally intact indicates that dementia is not an inevitable accompaniment of old age. Nonetheless, statistics back up plain life experiences in showing that the main risk factor for AD is old age. The risk for developing AD in old age can be assessed partially based on the ApoE genotype. The contribution of general health and the environment is difficult to factor in, and there may be other genetic risk factors that we don't know about. Possibly, whether a person with ApoE4/4 develops AD at 70 vs 75 years depends on general health and the environment. Stroke, CNS infections, traumatic brain injury, and any type of brain damage deplete structural and functional reserves and aggravate the dementia.
Old age without clinical dementia is associated with some loss of neurons and synapses and an overall reduction of brain weight by 200 gm. The remaining neurons are enough to carry out neurological function. Some compensatory dendritic sprouting is also seen. Neuronal plasticity (the ability to make new synapses) is enhanced by trophic factors (neurotrophins). The best known neurotrophin, nerve growth factor, is important for growth and maintenance of cholinergic neurons that are depleted in AD. Neuronal activity also enhances plasticity.
VASCULAR DISEASE IN AD
An important cause of the neurological dysfunction in AD is ischemia. This is caused by cerebral amyloid angiopathy (CAA), which is found in about 90% of AD cases, and cerebral atherosclerosis and small vessel disease, which is found in the majority of patients. Middle age hypertension is also a risk for AD. Soluble Aβ is a potent vasoconstrictor of cerebral vessels. Amyloid deposition on the vascular wall results in loss of smooth muscle and narrowing of the lumen. With the loss of their smooth muscle, cerebral arterioles lose their ability to constrict and dilate in response to regional brain activation. Narrowing of capillaries decreases cerebral perfusion and impairs the blood brain barrier function. Occluded capillaries form the seed of SPs. Thus, the brain capillary network is diminished and does not regenerate because of senescence. Other consequences of CAA are lobar hemorrhage, cerebral infarcts, and leukoencephlopathy. These changes are superimposed on cerebral atherosclerosis and its complications.
NEUROTRANSMITTERS IN AD
The brain, in AD, shows a loss of cholinergic neurons in the basal forebrain, decreased acetylcholine (Ach) levels, and a decrease in the acetylcholine synthesizing enzyme choline acetyltransferase (CHAT) in the cerebral cortex. Animal models show that Ach plays a crucial role in information processing and memory. Although other neurotransmitter systems (noradrenalin, serotonin, somatostatin and other peptides) are also deficient in AD, the cognitive impairment correlates best with the loss of cholinergic input. Acetylcholinesterase inhibitors (tacrine) and Ach receptor agonists, including nicotine, have been used to treat AD. The marginal success of this approach suggests that, in addition to Ach deficiency, there are other profound alterations that contribute to the cognitive dysfunction.
DIAGNOSIS OF AD
There are no specific clinical findings in AD. However, progressive dementia evolving over a few years without focal neurologic deficits or abnormal imaging findings is probably AD. Elevation of tau protein and decrease of Aβ in the CSF are useful biomarkers. A definitive diagnosis can only be made by pathological examination of brain tissue. One of the most frequent reasons for requesting an autopsy today is in order to establish the diagnosis of AD. Autopsy studies show that the brains of most people over 65, even without clinical dementia, contain a few SPs and NFTs in the hippocampus and entorrhinal cortex. This suggests that formation of SPs and NFTs is part of the aging process. The brains of demented people contain more SPs and NFTs, not only in the limbic cortex but also in the neocortex and other regions. The more numerous and widespread the SPs and NFTs, the more severe the dementia.
NEUROIMAGING MARKERS IN AD
Listed in the table below are imaging technologies that contribute to the diagnosis of AD. The abnormalities detected by these methods correlate with the autopsy findings in AD. Furthermore, studies using neuroimaging show that these abnormalities are progressive and their appearance precedes by several years the development of clinical symptoms of dementia.
| NEUROIMAGING TECHNOLOGY | FINDINGS-INTERPRETATION |
|---|---|
| PiB PET Fibrillar Aβ deposits are imaged in vivo by PET scan, following administration of 11C Pittsburgh Compound B (PiB), a substance that crosses the blood-brain barrier and binds selectively to Aβ. PiB binding detects Aβ in plaques and in blood vessels and gives an overall estimate of Aβ load. |
[11C] PiB PET matches the distribution and quantity of Aβ as seen at autopsy. Aβ is initially distributed diffusely through the neocortex and then spreads to the temoral lobe, hippocampus, and deep nuclei. |
| FDG-PET PET scan using [18F]fluorodeoxyglucose (FDG-PET) measures cerebral glucose metabolism and blood flow, which are indicators of synaptic function. |
In AD, there is decreased metabolism in the temporo-parietal cortex at resting state, reflecting a defect in episodic memory. |
| fMRI Functional MRI (fMRI) detects change in neuronal activity during the performance of cognitive tasks, by measuring blood-oxygen-level-dependent (strong) signal. |
In AD, there is decreased activation of the hippocampus during episodic memory tasks. |
| DTI Diffusion tensor imaging (DTI) detects axon density by measuring diffusivity of water melecules in the white matter. |
In AD, the white matter connecting to the hipocampus shows decreased density, probably due to neuronal loss. |
The finding of AD pathology in nondemented old people presents the neuropathologist with a dilemma. Should cases with a few SPs and NFTs be diagnosed as AD, even if there is no clinical dementia? The NIA-Reagan and CERAD diagnostic criteria for AD (see references below) take into account not only the numbers of SPs and NFTs but also age and intellectual function. Certainly the presence of SPs and NFTs indicates that the biochemical abnormalities that cause AD are set in motion. In some individuals, this process advances rapidly and causes severe dementia; in others, it is slow and causes forgetfulness and a mild decline in mental power. While old age is the most important risk factor for AD, it is worth emphasizing that 90% of people over 65 have no clinical dementia. Despite significant advances in the past 20 years, major gaps in our knowledge of AD remain. Knowledge about the neurotransmitter deficiencies, neuronal plasticity, and the role of the environment and body-brain interactions may provide a basis for designing treatment protocols for AD.
Further Reading
- Consensus recommendations for the postmortem diagnosis of Alzheimer's disease. The National Institute on Aging, and Reagan Institute Working Group on Diagnostic Criteria for the Neuropathological Assessment of Alzheimer's Disease. Neurobiol Aging. 1997;18(4 Suppl):S1-2. PubMed
- Mirra SS, Heyman A, McKeel D, et al. The Consortium to Establish a Registry for Alzheimer's Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer's disease. Neurology. 1991;41:479-86. PubMed
- Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol (Berl). 1991;1991;82:239-59. PubMed
- Goedert M, Spillantini MG. A century of Alzheimer's disease. Science 2006;314:777-81. PubMed
- Bacscai BJ, Frosch MP, Freeman SH, et al. Molecular imaging with Pittsburgh Compound B confirmed at autopsy. A case report. Arch Neurol 2007;64:431-34. PubMed
- Castellani RJ, Lee HG, Zhu XJ, et al. Alzheimer Disease Pathology As a Host Response. J Neuropathol Exp Neurol. 2008;67:523-31. PubMed
- Nelson PT, Braak H, Markesbery WR. Neuropathology and Cognitive Impairment in Alzheimer Disease: a Complex but Coherent Relationship. J Neuropathol Exp Neurol. 2009;68:1-14. PubMed
- Avramopoulos D. Genetics of Alzheimer's disease: recent advances. Genome Med 2009;1:34 PubMed
- Querfurth HW, LaFerla FM. Alzheimer's Disease. N Engl J Med 2010;362:392-44. PubMed
- Kroner Z. The relationhip between Alzheimer's Disease and Diabetes: Type 3 Diabetes? Altern Med Rev 2009;14:373-9. PubMed
- Ewers M, Sperling RA, Klunk WE, Weiner MW, Hampel H. Neuroimaging markers for the prediction and early diagnosis of Alzheimer's disease dementia. Trends Neurosci 2011;34:430-42. PubMed
- Liu L, Drouet V, Wu JW, Witter MP, Small SA, Clelland C, Duff K. Trans-synaptic spread of tau pathology in vivo. PLoS One. 2012;7(2):e31302. Epub 2012 Feb 1. PubMed
- Nelson PT, et al. Correlation of Alzheimer Disease Neuropathologic Changes with Cognitive Status:A Review of the Literature. J Neuropathol Exp Neurol 2012:71:362-81. PubMed
- Hyman BT et al. National Institute on Aging–Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease. Alzheimer's & Dementia 2012;8: 1–13. PubMed
- Alzheimer's Association, 2012 Alzheimer's Disease Facts and Figures, Alzheimer's & Dementia, Volume 8, Issue 2
- Mhatre SD, Tsai CA, Rubin AJ, et al. Microglial Malfunction: The Third Rail in the Development of Alzheimer's Disease. Trends in Neurosciences 2015;38:621-36.
- DeTure MA, Dickson DW. The Neuropathological Diagnosis of Alzheimer's Disease. Mol Neurodegener 2019 Aug 2;14(1):32. PubMed
Updated: April, 2020