NORMAL ANATOMY
Myofibers are long syncytial cells with thousands of nuclei. They measure from 10 to 100 microns in diameter and from a few mm to 3 cm or more in length. In cross-sections, myofibers are normally polygonal. In longitudinal sections, they display cross striations. Their nuclei are located under the sarcolemma, except near myotendinous junctions, where myofibers become narrower and myonuclei are central. Myofibers are attached to fasciae and aponeuroses and are anchored to bones by tendons. Each muscle is wrapped by a fibrocollagenous sheath, the perimysium. Fibrovascular septa projecting from this sheath divide the muscle into fascicles of up to 100 fibers. The endomysium is a space between individual muscle fibers that carries capillaries. It contains a minimal amount of matrix including collagen. In muscular dystrophies and other wasting diseases of muscle, endomysial fibrous and adipose tissue increase and replace lost muscle. Endomysial fibrosis causes muscles to become stiff.
Myofibers contain two kinds of contractile filaments: myosin: 12-15 nm thick - 1.5 micron long, and actin: 6-7 nm thick - 1 micron long. Actin filaments are attached to Z- bands which are composed of actinin and other proteins. The Z-bands and the overlap of thin and thick filaments give muscle its cross-striated appearance. Sarcomere is the distance between two successive Z-bands. During contraction, actin and myosin filaments slide into each other and the sarcomere becomes shorter by 20%. The contractile filaments in each myofiber are arranged in 1-2 micron thick bundles, (myofibrils), which are visible with the light microscope. Myofibers contain also intermediate cytoskeletal filaments between myofibrils. These measure 7-12 nm and are composed mainly of desmin and vimentin.
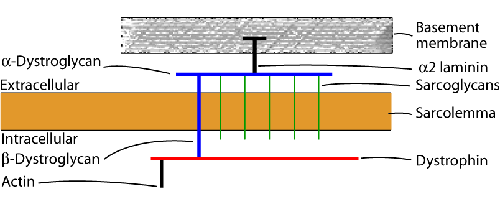 |
The contractile part of myofibers is bound to the extracellular matrix by a chain of proteins (see diagram on left), the main components of which are dystrophin and the dystrophin-associated complex (DAC). Dystrophin, a 427-kD protein, lies under the sarcolemma (the cell membrane of muscle) and is attached to cytoskeletal actin by its C-terminus. The DAC consists of two main complexes of transmembrane proteins, the dystroglycans (two subunits) and the sarcoglycans (five proteins), which are closely associated with one another. The N-terminus of dystrophin is bound to b-dystroglycan. Dystroglycan, in turn, is bound to merosin, the alpha 2-chain of the basement membrane protein laminin 2. Defects in this chain cause progressive muscle damage resulting in muscular dystrophy. Mutations of dystrophin are the basis of Duchenne and Becker muscular dystrophies. Defects of the sarcoglycan complex cause some of the limb girdle dystrophies. Deficiency of merosin is found in some congenital muscular dystrophies. Antibodies to these proteins applied to sections of muscle are used to diagnose muscular dystrophies.
 ATPase histochemical stain |
 NADH histochemical stain |
Histochemical staining of frozen muscle using myofibrillar ATPase and oxidative enzyme stains shows two fiber types, type 1 (slow-red) and type 2 (fast-white). These fibers have distinct myosin isoforms. Type 1 fibers are rich in oxidative enzymes, mitochondria, myoglobin (an oxygen carrier), and lipid. They are capable of protracted slow action and clonic activity. Type 2 fibers are rich in glycogen and glycolytic enzymes. They are capable of fast, powerful, tonic contraction. By varying the pH of the ATPase preincubation, type 2 fibers can be further subdivided into 2a and 2b. Fiber types are segregated in birds (red and white meat). In mammals, type 1, 2a, and 2b fibers are present in all muscles, mixed like a checkerboard. Their proportion varies in different muscles.
The immediate source of energy for muscle contraction is hydrolysis of ATP. Anaerobic glycolysis and oxidative phosphorylation replenish ATP. Muscle has another source of energy also, phosphocreatine. Energy in phophocreatine cannot be used directly for muscle contraction, but is used to replenish ATP by transferring a high energy phosphate to ADP. When ATP is plentiful, it transfers the phosphate back to creatine. Thus, phosphocreatine serves as a reserve and buffer for ATP. The enzyme creatine kinase (CK), that moves phosphate from creatine phosphate to ADP, is present in abundance in muscle. When the sarcolemma is damaged, CK leaks out into the serum. Serum CK is a biochemical marker of myonecrosis. In type 2b fibers, energy for restoration of ATP and creatine phosphate, is derived primarily from anaerobic glycolysis. Type 1 fibers derive most of their energy from oxidative phosphorylation. Type 2a fibers are intermediate between type 1 and type 2b.
The motor unit is a group of 100-200 myofibers, all innervated by the same motor neuron. These fibers are not aggregated, but are scattered over a radius of a few millimeters and overlap with myofibers of adjacent motor units. All fibers of a motor unit are of the same histochemical type, i.e. either type 1 or type 2. Thus, the histochemical type of a given myofiber is determined by its nerve supply. If that changes, the histochemical type will change accordingly.
APPROACH FOR EVALUATION OF MUSCLE DISORDERS
When a patient presents
with muscle weakness, the first step should be to
determine if this is a manifestation of systemic
illness or a neuromuscular disorder. The clinical
evaluation should focus on the pattern of weakness
(proximal vs distal), fatiguability, soreness, skin
rash, muscle atrophy, and fasciculations. Family
history is important. Useful laboratory studies
include CK, erythrocyte sedimentation rate, serology
for collagen vascular disease, acetylcholine receptor antibodies, and electromyography
(EMG). Functional
testing (Tensilon test) are diagnostic in some
cases.
Based on the clinical
and laboratory evaluation, muscle disease can be
divided into two broad categories, myopathy and
denervation atrophy. Myopathy (primary
disease of muscle) to a clinician means a condition
with proximal weakness, elevated CK, and myopathic
EMG changes. The latter consist of low voltage and
short motor unit potentials caused by depletion
of myofibers of the motor unit. This broad group
includes the muscular dystrophies and inflammatory
myopathies. Denervation most
often causes distal weakness and atrophy, and neuropathic
EMG changes (see further on). CK is normal.
A more specific diagnosis can be obtained by a muscle
biopsy which
should be used as a last resort.
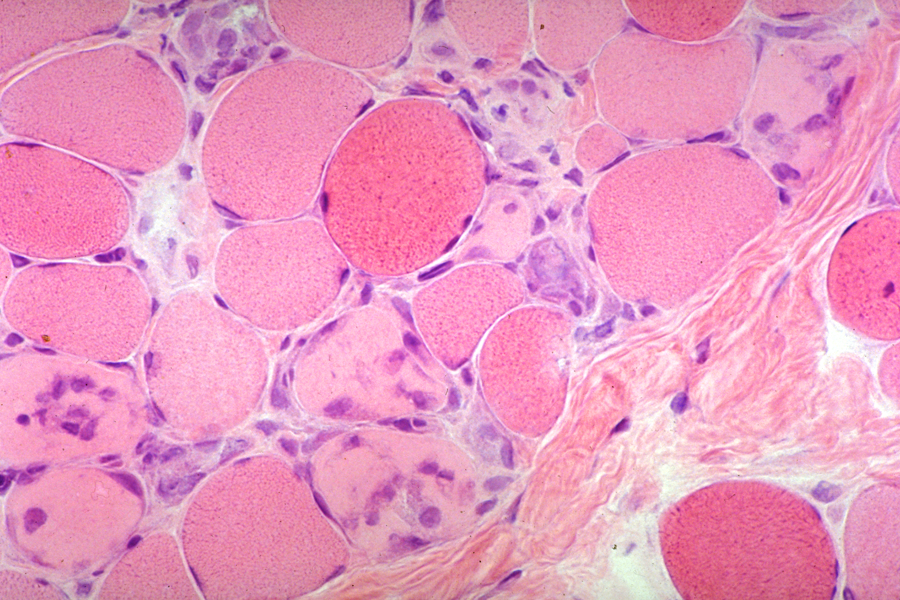

Muscle has a limited repertoire of reactions to injury which consists basically of atrophy and myonecrosis. Myonecrosis is segmental. Necrotic fibers are invaded by macrophages which remove the debris. Myonuclei from adjacent intact segments proliferate and initiate regeneration. Regenerating fibers are slender, basophilic, and have central rows of large nuclei. Necrosis and regeneration go hand in hand but eventually one or the other prevails, depending on the disease. Myonecrosis occuring over a long period, as in the muscular dystrophies, results in loss of muscle. Remaining myofibers are often hypertrophic and have structural abnormalities such as internal nuclei, internal splits, and an irregular orientation of their myofilaments. Lost muscle is replaced by adipose tissue and collagen. To the pathologist, myopathy is a muscle disease with myonecrosis and structural abnormalities. Inflammatory myopathies are characterized by myofiber necrosis and inflammation. Denervation causes mainly atrophy and a change in the pattern of histochemical staining (see Denervation).