WERNICKE-KORSAKOFF SYNDROME
The Wernicke-Korsakoff syndrome (WKS) is a combination of oculomotor abnormalities and mental symptoms seen in malnourished patients with vitamin B1 (thiamine) deficiency. It is most common in alcoholics, but affects also demented people who neglect their nutrition, patients with gastric cancer, persons who have had bariatric surgery, gastrectomy (sometimes appearing many years after the procedure), hyperemesis gravidarum, anorexia nervosa, and other malnutrition settings. The WKS has also been reported following treatment with fedratinib, an inhibitor of thiamine uptake and transport, and other chemotherapeutic agents. The syndrome has 2 components: Wernicke�s encephalopathy (WE), and Korsakoff�s amnesia (KA). The classic clinical presentation of WE is a triad of eye abnormalities (nystagmus, oculomotor paralysis, paralysis of conjugate gaze), ataxia of stance and gait, and mental symptoms, such as withdrawal and confusion. This may occur alone be accompanied by KA which is characterized by anterograde and retrograde amnesia and a distortion of memory such that patients recount imaginary stories that may have some basis in fact. An axonal peripheral neuropathy (a form of Beri-Beri disease) affecting the distal parts of the extremities is seen in 80% of patients. The syndrome may be partial, and a high index of suspicion is essential for diagnosis, especially in debilitated patients with unexplained confusion. The neurological symptoms may be accompanied by high output cardiac failure with peripheral edema. Administration of thiamine causes a rapid reversal of ophthalmoplegia within hours. Recovery from nystagmus and confusion is slower. Amnesia does not respond to treatment fully.
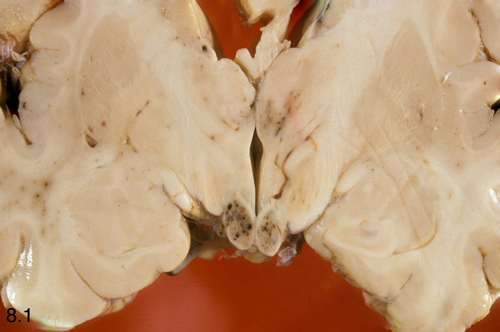 Acute WKS: Mammillary body hemorrhages |
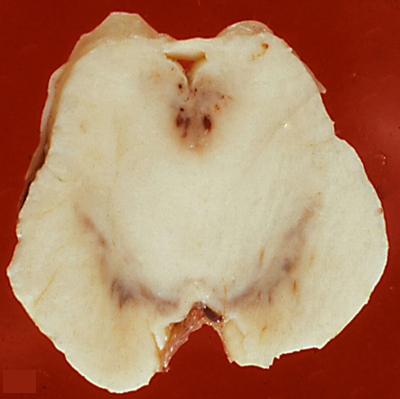 WKS, acute lesion. Periaqueductal lesions. |
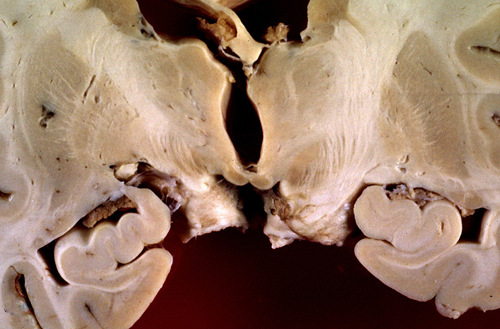 WKS, burned out lesion. |
The histopathology of WKS is incomplete loss of neurons, damage of axons and myelin which casuses loosening or vacuolization of the neuropil, and punctate hemorrhages.The lesions have a characteristic anatomical distribution which includes the mammillary bodies, the hypothalamus, thalamus, periaqueductal gray matter, colliculi, and the floor of the fourth ventricle. The mammillary bodies are involved in all cases. This is the hallmark of WKS. Lesions of the colliculi and the floor of the fourth ventricle (oculomotor nuclei, vestibular nuclei, dorsal motor nuclei of the vagus) cause the oculomotorand brain stem signs. Involvement of the medial dorsal nuclei of the thalamus was initially thought to be responsible for the memory defect, but more recent studies have found that this correlates with lesions of the mammilothalamic tract and anterior thalamus. Anterograde amnesia has been reported in thalamic infarcts involving the mammillothalamic tracts (which project to the anterior thalamic nuclei). An identical memory defect (Korsakoff's amnesia) is caused by bilateral hippocampal damage. The hippocampus projects to the mammillary bodies via the fornix but is not affected in the WKS. The MRI shows a hyperintense FLAIR signal in the affected areas. In full-blown WKS, all these structures are involved. In less severe cases, some may be spared. Each successive bout of WKS causes additional loss of neural tissue. Old cases show atrophy of the mammillary bodies and dilatation of the third ventricle. In 30% to 50% of cases, the cerebellum shows degeneration (neuronal loss) of the superior vermis (see below).
The WKS is due to thiamine (B1)deficiency. Thiamine pyrophosphate, the active form of thiamine, is required for oxidative metabolism of pyruvate for ATP production. The topography of the lesions in the WKS resembles the Leigh syndrome, a metabolic disorder caused by defects of the pyruvate dehydrogenase complex and the respiratory chain. This suggests that damage of sensitive CNS structures in the WKS is due to energy failure. Thiamine is stored in several organs (heart, kidneys, liver, brain, muscles). These stores can be depleted in malnourished patients. Alcohol displaces more nutritious foods from the diet, adds carbohydrates that use up thiamine, and impairs the absorption of vitamins. Intravenous glucose administration to a patient with borderline thiamine deficiency may trigger the WKS. Therefore, vitamin B1 supplementation is mandatory for patients at risk.
MIDLINE CEREBELLAR DEGENERATION
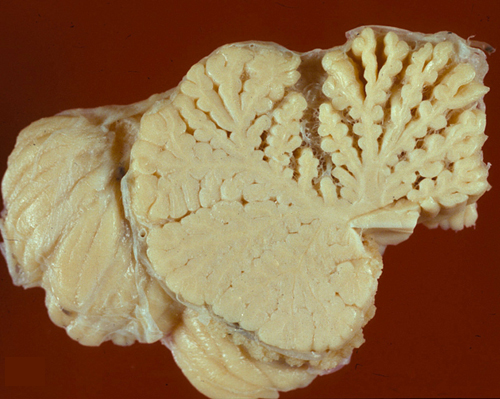 Midline cerebellar degeneration |
Midline cerebellar degeneration (also referred to as alcoholic cerebellar degeneration) is a component of WKS, but may also occur alone. It causes ataxia of stance and gait with relative sparing of the arms. It has an insidious onset and a subacute or chronic course. Pathologically, there is loss of Purkinje and granular neurons. In most cases, the lesions are confined to the superior vermis, which is grossly atrophic.
In severe cases, degeneration spreads to the cerebellar hemispheres, causing ataxia of leg movements. Localization of the pathology in the superior vermis distinguishes this entity from other familial and sporadic cerebellar degenerations.
VITAMIN E DEFICIENCY
Deficiency of the lipid-soluble vitamin E occurs in cases of intestinal malabsorption such as cystic fibrosis, congenital biliary atresia, intestinal resection, and abetalipoproteinemia (Bassen-Kornzweig syndrome). Clinically, vitamin E deficiency causes a sensory peripheral neuropathy, ataxia, retinitis pigmentosa, and skeletal and cardiac myopathy. Neuropathological examination in such cases reveals loss of dorsal ganglionic neurons with degeneration of their peripheral and central axons (peripheral neuropathy and posterior column degeneration respectively). Neuroaxonal swellings are seen in the gracile and cuneate nuclei. Similar changes can be produced in rats and monkeys with experimental vitamin E deprivation. The neurodegenerative effects of vitamin E deficiency are due to loss of the anti-oxidant action of alpha tocopherolthe active form of vitamin E, and can be prevented with vitamin E supplementation.
VITAMIN B12 DEFICIENCY
The main dietary sources of vitamin B12 (cobalamin) are animal products, such as meat and dairy foods. In the stomach, cobalamine is bound to intrinsic factor, a glycoprotein produced by the parietal cells of the stomach. The cobalamine-intrinsic factor complex is transported to the terminal ileum, where it binds to receptors on the brush border of enterocytes and is absorbed. The common setting of vitamin B12 deficiency is pernicious anemia, an autoimmune disorder caused by antibodies against gastric parietal cells and the intrinsic factor. Vitamin B12 deficiency may also result from Helicobacter pylori gastritis, surgical resection, tumors, and other conditions involving large parts of the stomach or the lower ileum. Strict vegetarians (vegans) who omit all animal food from their diet may also be vitamin B12 deficient. Nutritional vitamin B12 deficiency in breast-fed infants of vegan mothers causes irritability, lethargy, failure to thrive, neurodevelopmental regression, and involuntary movements referred to as the "infantile tremor syndrome". Treatment with vitamin B12 rapidly reverses these abnormalities. Nitrous oxide-NO (laughing gas) inactivates vitamin B12 and its recreational use may cause functional vitamin B12 deficiency.
Lack of vitamin B12 produces hematologic abnormalities (megaloblastic anemia) and neurologic complications (subacute combined degeneration of the spinal cord-SCD). The hematologic abnormalities are probably due to a disorder of DNA synthesis. Megaloblastic anemia can be corrected by administration of folate alone. The neuropathological abnormalities are due to a different, as yet unknown, biochemical mechanism. Inherited metabolic disorders of cobalamin-dependent enzymes do not cause SCD. It has been suggested that cobalamin plays a role in the expression of tumor necrosis factor alpha (TNF-a) and epidermal growth factor (EGF), which is separate from its catalytic activity. In cobalamin deficiency, TNF-a is upregulated and EGF downregulated. This imbalance may account for the damage of the white matter.
At first, SCD causes weakness and paresthesias of the hands and feet. As the pathology advances, vibration and position sense are lost and the gait becomes ataxic. Weakness gets worse and spasticity of the limbs appears. The advanced state is characterized by spastic paraplegia, contractures, ataxia, and impairment of other sensory modalities.
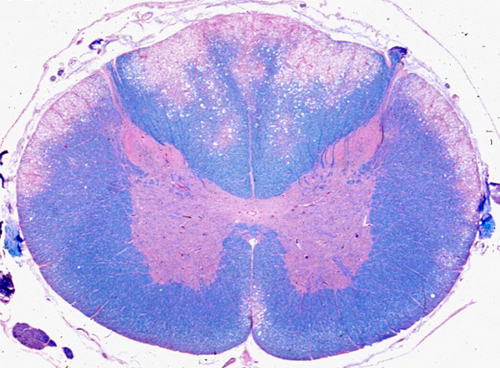 Subacute combined degeneration |
The earliest neuropathologic lesion is distention of myelin sheaths, imparting a spongy appearance to the affected white matter. This is followed by disintegration of myelin, which is removed by macrophages. Loss of axons also occurs, but is less severe than the loss of myelin. The lesions affect initially the posterior and lateral (combined) columns of the upper thoracic and low cervical spinal cord. They do not affect anatomical fiber systems, but rather involve the white matter in a symmetric nonselective fashion. In advanced cases, the entire circumference of the spinal cord is affected and the pathology extends to peripheral nerves (myeloneuropathy). The optic nerves are rarely involved. All these changes can be prevented (and in their early stages, reversed) by vitamin B12 administration.
Further Reading
- Renthal W, Marin-Valencia I, Evans PA. Thiamine Deficiency Secondary to Anorexia Nervosa: An Uncommon Cause of Peripheral Neuropathy and Wernicke Encephalopathy in Adolescence. Pediatr Neurol 2014; 51: 100-103. PubMed
- Scalabrino G, Peracchi M. New insights into the pathophysiology of cobalamin deficiency. Trends Mol Med. 2006;12:247-54.PubMed
- Goraya JC, Kaur S, Mehra B. Neurology of Nutritional Vitamin B12 Deficiency in Infants: Case Series From India and Literature Review. J Child Neurol 2015; May 7. PubMed
Updated: May, 2023