GENERAL PRINCIPLES
 Bullet track |
Some
traumatic lesions, such as epidural and
subdural hematomas, merely compress
the brain and raise the intracranial
pressure. Other lesions, such as contusions
and diffuse axonal injury, cause structural
brain damage.
In addition
to mechanical injury, trauma induces
also complex neurochemical alterations
that disturb cerebral blood flow, increase
vascular permeability, and produce beta
amyloid precursor protein. Structural
lesions induce neuroinflammation. These
secondary changes compound the mechanical
effects. The most important secondary
processes in TBI are HIE and increased
intracranial pressure. HIE
and focal ischemic lesions are
caused by cardiorespiratory arrest occurring
in severe concussion, the release of
excitatory neurotransmitters by neurons,
vascular spasm in cases with subarachnoid
hemorrhage, direct traumatic vascular
disruption, and other factors.
In severe
TBI, there are often multiple lesions
and different types of lesions, e.g.,
epidural hematoma, subdural hematoma,
contusions, etc. Even if there are no
detectable lesions, head injury can cause
transient loss of neurological function
and autonomic paralysis (cerebral concussion).
When a patient with severe TBI arrrives
at the Emergency Department, the Glascow
Coma Scale can give a rough idea of the
severity of neurological depression but
it is hard to sort through the effects
of different lesions and predict if neurological
function will be restored. Mortality,
in the immediate post traumatic period,
is due mainly to increased
intracranial pressure and HIE. Survivors from severe
TBI may have seizures, focal neurologic
deficits, dementia, or the persistent
vegetative state.
Trauma is a risk factor for Alzheimer's
disease.
SKULL FRACTURES
A skull fracture is classified by the configuration or pattern it displays. The most frequent type, linear fracture, is a straight crack or break produced by a blow to the skull. A fracture that is displaced by a distance equal to the thickness of the skull or more is a depressed skull fracture. A skull fracture does not necessarily indicate underlying brain damage. Skull fractures may create a communication between the intracranial compartment and septic areas such as air sinuses, nasal fossae, and middle and external ear, leading to infection of the brain and meninges.
EPIDURAL HEMATOMA
 Epidural hematoma |
 Epidural hematoma |
 Epidural hematoma-CT |
There is no epidural space normally in the cranium. The dura is adherent to the skull. Fracture of the inner table of the skull can tear arteries and veins that run between the dura and the skull. A blow to the head may cause instant deformation of the skull without a fracture. This, too, can cause vascular tears. Bleeding from these vessels lifts the dura off of the skull forming an epidural blood clot. Epidural hematomas develop most commonly with fractures of the squamous portions of the temporal and parietal bones that tear the middle meningeal vessels. Less commonly, they result form tears of diploic veins and dural sinuses. Epidural hematoma is seen in 2.7% to 4% of TBI and has an overall mortality of 10%. Cranial fractures are present in 70% to 90% of cases. Symptoms of increased intracranial pressure in epidural hematomas with arterial rupture usually develop within hours after the injury. With venous bleeding, they take longer. There is a natural epidural space around the spinal cord. Spinal epidural hematoma may occur as a result of trauma, but may also develop spontaneously in patients with bleeding disorders.
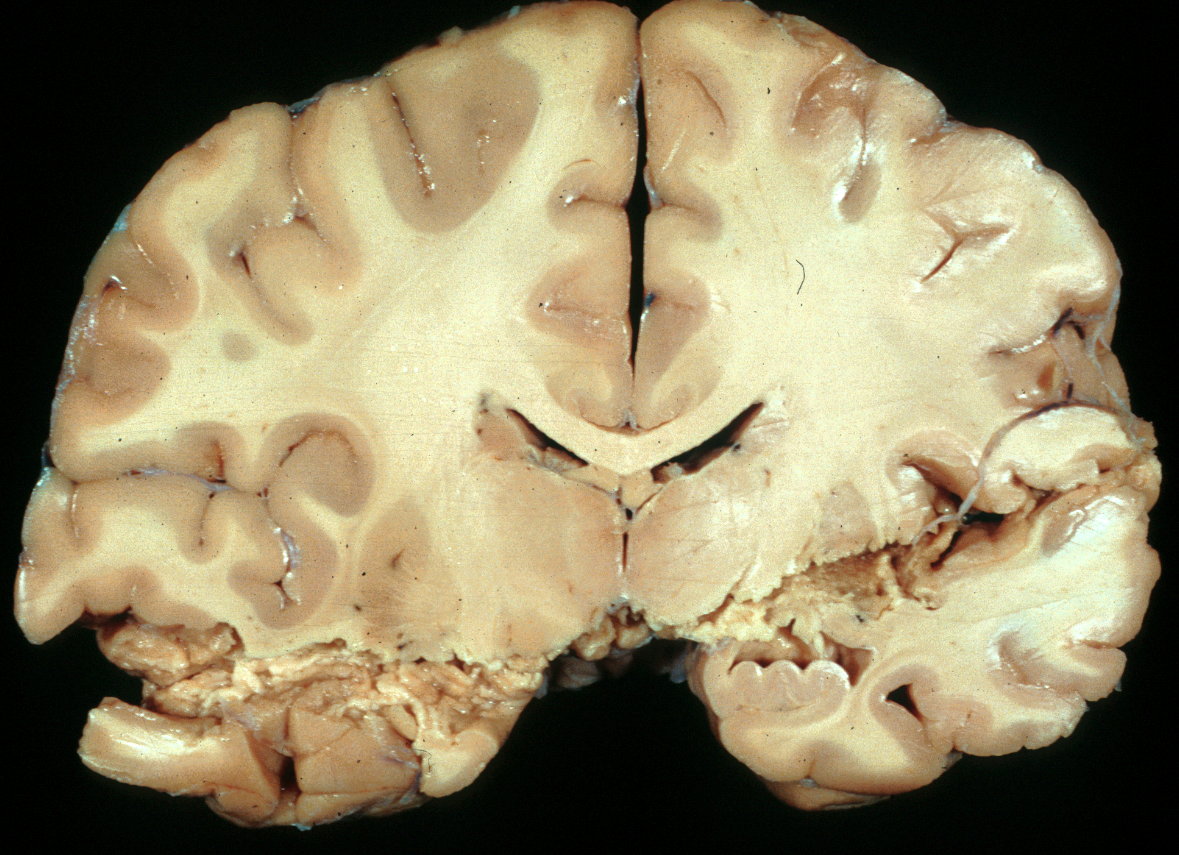
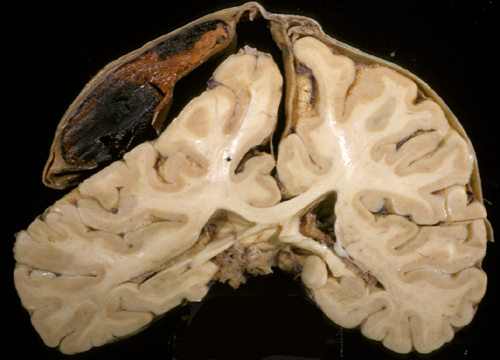
SUBDURAL HEMATOMA
 Subdural hematoma |
Acute subdural hematoma (ASD) is seen in 12% to 29% of severe TBI and and has a mortality rate of 40% to 60%. Some ASDs are caused by blood from hemorrhagic contusions and traumatic subarachnoid hemorrhage that extends to the subdural space due to tears of the arachnoid membrane. In other cases, ASDs are caused by rupture of bridging (emissary) veins, which run between the surface of the brain and the skull and are especially numerous along the superior sagittal sinus. Excessive movement of the brain causes rupture of these vessels, which are attached to the skull. Individuals with brain atrophy, in whom the bridging veins are stretched and there is more room for the brain to move, are especially prone to developing subdural hematoma. Such ASDs may occur with mild or trivial head trauma. The same thing may happen in patients with hydrocephalus, if the ventricles collapse rapidly after shunting. Less commonly, subdural hematomas result from rupture of arteries that accompany bridging veins.
Large subdural hematomas raise the intracranial pressure and compress the brain. With arterial bleeding, symptoms develop rapidly. In many instances, especially with venous subdurals of infants and old people, there is an interval between trauma and the onset of symptoms. Sometimes the preceding injury is insignificant, or no history of trauma can be elicited.
 Organizing subdural hematoma |
The subdural hematoma starts as a flat blood clot between the dura and the arachnoid membrane. Initially, it is not attached to the dura. Fibroblasts, growing from the dura into the clot, organize it. In 5 to 6 days, fibroblast growth causes the blood clot to be loosely attached to the dura. In 10 to 20 days, a loose fibrous membrane is formed between the dura and the clot (outer membrane). Fibrous tissue then grows around the edges of the hematoma and along its inner surface (inner membrane), encapsulating it completely. Maturation of connective tissue results, after several weeks or months, in formation of a sac with a fibrous wall (chronic subdural hematoma). Blood in this sac is absorbed to a variable degree, and the cavity contains clear or hemorrhagic fluid and a loose, vascular connective tissue. Rupture of delicate vessels may cause repeated bleeding in the sac. Fluid may also leak into the cavity from immature capillaries. If a large amount of CSF enters the subdural space during the traumatic event, it washes off the blood, and no clotting or organization takes place. The histological appearance of the sac is helpful in estimating the duration of the subdural hematoma.
Subdural hematomas in infants are sometimes accompanied by brain swelling, hypodensity, and loss of gray-white matter differentiation in the underlying hemisphere. This combination has been called "the big black brain" and is associated with a fatal outcome or severe brain damage in survivors. The pathogenesis of this lesion is not completely understoood but it probably has to do with hypoperfusion of the hemisphere underlying the subdural in combination with systemic insults, such as apnea and seizures. The involved hemisphere in survivors shows severe atrophy and neuronal loss and gliosis in the cortex, basal ganglia and thalamus, consistent with unilateral HIE changes.
Subdural hygromas are fluid collections seen most commonly in very young and very old individuals. On imaging, they have a higher density than CSF and contain fluid with a high protein content or with blood degradation products. The pathogenesis of subdural hygromas is uncertain and their clinical course unpredictable. Some evolve from chronic subdurals but most are diagnosed incidentally when patients are having imaging studies for other reasons. Some subdural hygromas recede spontaneously and others remain for variable periods of time.
SUBARACHNOID HEMORRHAGE
 Subarachnoid hemorrhage. |
Subarachnoid hemorrhage is the most frequent traumatic brain lesion. It results from rupture of corticomeningeal vessels and from hemorrhagic contusions of the brain. Usually it is diffuse and does not exert localized pressure. Blood is diluted by the CSF and does not clot unless it is massive. A large subarachnoid hemorrhage raises the intracranial pressure, impairs cerebral perfusion and causes HIE. Hemoglobin released form RBCs in the subarachnoid space triggers vascular spasm. It also incites fibrosis of the arachnoid membrane and the subarachnoid space, which may impair CSF circulation leading to hydrocephalus.
Further Reading
- Duhaime AC, Durham S.Traumatic brain injury in infants: the phenomenon of subdural hemorrhage with hemispheric hypodensity ("Big Black Brain").Prog Brain Res. 2007;161:293-302.PubMed
Updated:February, 2017