Hypoxic - Ischemic Encephalopathy. General Principles
The brain is about 2% of the total body mass but consumes 15% of the energy generated in the body, more than any other organ, including muscle. The brain regulates energy allocation to muscle and other organs but gives priority to its own energy needs. Energy consumption is highest during mental activity and sensory stimulation but does not cease during sleep. Most of this energy is used by neurons to maintain resting potentials and restore ionic concentrations after synaptic transmission. Some energy is also used to support synthetic and catabolic activity of neurons and glial cells and, in young age, for growth.
Energy for these functions is derived from hydrolysis of adenosine triphosphate (ATP). Thus, the brain is like a chemical battery of ATP which must be constantly recharged. Unlike some other tissues, brain cells have no backup energy source such as creatine phosphate. The brain has no energy stores of its own except for a small amount of glycogen in astrocytes. Anaerobic glycolysis of this glycogen provides neurons with some lactate that can be used by mitochondria but is insufficient to meet energy needs. The brain can also use lactate from the circulation.
Fatty acids cannot be used because they are not transported across brain capillaries, but the brain can use ketone bodies derived from fat. In cases where the ketogenic diet is employed to manage drug-resistant epilepsy, ketone bodies serve as the principal energy source. However, the bulk of ATP is derived from oxidative phosphorylation of glucose. Thus, the brain depends on a second by second supply of oxygen and glucose by the blood.
Glucose does not diffuse freely across the blood-brain barrier. It is transported by specialized transporters located in the end feet of astrocytes that surround cerebral capillaries. A drop in cerebral perfusion, hypoxia, hypoglycemia, and severe anemia can cause a critical shortage of energy (energy crisis). In protracted generalized seizures, neurons use up glucose and oxygen faster than they are supplied, and discharge glutamate (see below) with the same result.
The most common cause of energy crisis is a drop of cerebral perfusion (global ischemia), usually resulting from cardiac arrest or severe hypotension (shock). Sustained severe hypoglycemia, and seizures lasting 1-2 hours also cause permanent brain damage. Pure hypoxia in clinical settings is unusual. Patients with lung disease are treated with oxygen and the brain can adapt to pure hypoxia, especially if it develops slowly. Hypoxia develops acutely in CO poisoning, which displaces oxygen from hemoglobin. Global ischemia is worse than hypoxia, hypoglycemia, and seizures because, in addition to causing energy failure, it results in accumulation of lactic acid and other toxic metabolites that are normally removed by the circulation.
The mechanism of neuronal damage in hypoxic-ischemic encephalopathy (HIE) is now beginning to be understood. Obviously, lack of energy causes initially electrical failure and, if it lasts long enough, results in arrest of cellular functions and cell death. However, animal experiments and clinical studies show that there are other factors, in addition to energy loss, that account for neuronal damage. Even sublethal HIE can set in motion a series of toxic reactions that finish off injured neurons and kill additional ones that have not been damaged during the initial insult. Thus, following global ischemia, neurons do not die suddenly or all at once. In some of them, damage develops hours or days after the insult. Most neurons undergo necrosis. In some neurons, HIE triggers apoptosis.
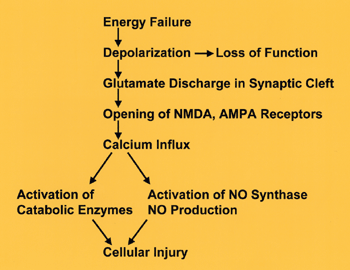
The first result of energy depletion is failure of Na+ and K+ pumps, leading to depolarization of the neuronal membrane (see diagram on left). Synaptic function and conductivity cease at this point. Depolarization causes neurons to release glutamate into the synaptic cleft. Glutamate is the most common excitatory neurotransmitter. In small amounts, it is indispensable for neuronal function. In excessive amounts, it is a neuronal poison, a toxin, and has been called excitotoxin. Some glutamate receptors are non-selective cation-permeable ion channels. Initially, over-activation of these channels causes a passive influx of Cl- (and Na+) into cells causing osmotic (cytotoxic) edema and rapid death. Additional structural damage develops hours or days later as a result of Ca++ influx into neurons. The NMDA and AMPA receptors of glutamate are channels that are permeable to Ca++. Activation of these receptors by excess glutamate causes massive influx of Ca++ into neurons. Ca++ activates catabolic enzymes (proteases, phospholipases, endonucleases). Ca++ also activates NO synthase, resulting in formation of the free radical NO. Additional free radicals result from the impairment of oxidative phosphorylation. Free radicals and activated catabolic enzymes destroy structural proteins, membrane lipids, nucleic acids, and other cellular contents, causing neuronal necrosis. DNA damage from endonucleases and mitochondrial injury from free radicals trigger apoptosis. Counteracting the action of glutamate is the basis of neuroprotective strategies that are now at an experimental stage.
Incomplete combustion of glucose results in lactic acidosis. Lactic acid can get through cell membranes and can damage not only neurons but glial and mesenchymal cells as well. Additionally, lactic acid and hydrogen ions cause cerebral edema by attracting water. Obviously, lactic acidosis is more severe in patients with HIE who are hyperglycemic and is not a significant factor in hypoglycemic encephalopathy or seizures.
Severe HIE is accompanied by cerebral edema and its effects are compounded by increased intracranial pressure. Cytotoxic (intracellular) edema develops in the initial phase of the insult. Interstitial cerebral edema, which follows, is due to vascular injury and the release in the interstitial space of vasoactive metabolites such as arachidonic and other fatty acids (derived from membrane glycerolipids), lactic acid, electrolytes and other unknown osmoles. Arachidonic acid also has a chemotactic function and induces acute inflammation.
Free radicals, lactic acid, cerebral edema, and inflammation cannot develop in unperfused, completely ischemic tissue. They develop following reperfusion. In an ironic sense, the brain has to be alive in order for the changes of neuronal death to develop. Thus, reperfusion is a double-edged sword. Without it, there is no hope for recovery. On the other hand, reperfusion causes additional (delayed) neuronal injury, brings monocytes to the site of injury, and sustains the glial and vascular reactions that follow. Knowledge of the various aspects of HIE and reperfusion injury may open the way to possible neuroprotective interventions.
THE PATHOLOGY OF HYPOXIC-ISCHEMIC ENCEPHALOPATHY
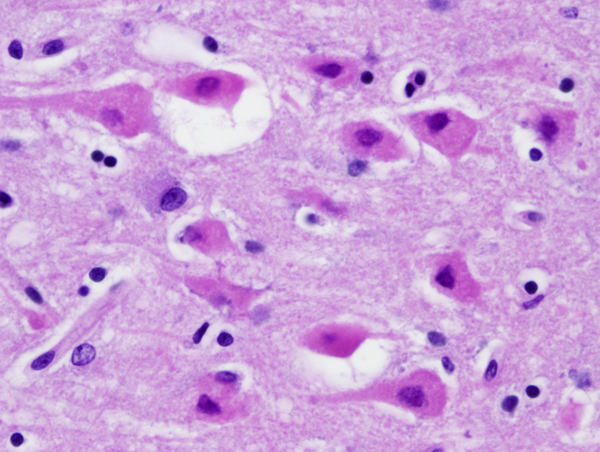 Anoxic neurons |
 Neuronophagia |
 Karyorrhexis |
With this background, let us examine what happens with different grades of HIE. Suppose that someone has a brief episode of global ischemia, say from fainting. Within seconds, energy failure causes electrical activity in neurons to cease and the patient loses consciousness. Neurons and glial cells are viable and, if circulation is promptly restored, the patient returns to normal. If, however, ischemia lasts longer, first the integrity of cell membranes will be compromised and then cellular metabolism will cease and neurons will die. Ischemia lasting 4-5 minutes can damage irreversibly hippocampal and neocortical pyramidal cells, striatal neurons, and Purkinje cells. More protracted ischemia can damage thalamic and brainstem neurons.
If a patient dies shortly after the insult, the brain is usually grossly and microscopically normal. If the patient survives and perfusion is restored, changes begin to appear within hours. At first, injured neurons shrink and become eosinophilic. This is due to increased density of damaged mitochondria. Neuronal nuclei condense. The shrunken eosinophilic neuron (anoxic neuron) is the hallmark of HIE. Astrocytes swell (Alzheimer type II cells). This is a poorly understood response of astrocytes to metabolic insults. If the patient survives longer, damaged neurons disintegrate and are removed by macrophages. With time, cortical atrophy and gliosis develop. In neonates and fetuses with HIE the condensed neuronal nuclei fragment, a phenomenon called karyorrhexis.
Some cases of HIE, usually after brief insults, cause neuronal death only without damage of glial cells (selective neuronal necrosis). Neurons are more sensitive than glial cells because they have higher energy demands and only they produce glutamate. Some neurons are more vulnerable than others (selective vulnerability). The hippocampal pyramidal cells of CA1, pyramidal neocortical neurons (layers 3, 5, and 6), Purkinje cells, and striatal neurons have the highest vulnerability.
 Hippocampal neuronal loss (right) post-HIE |
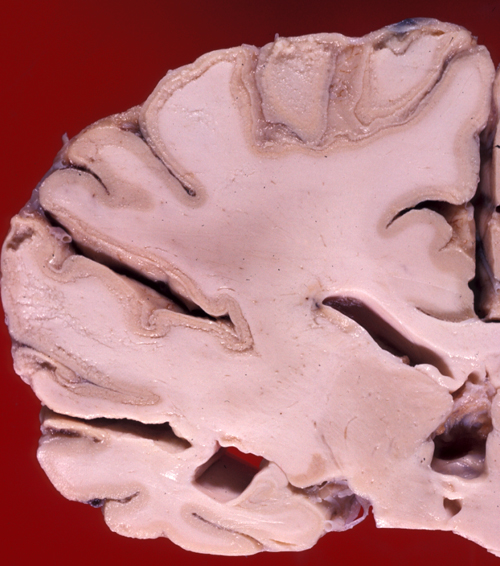 Pseudolaminar necrosis |
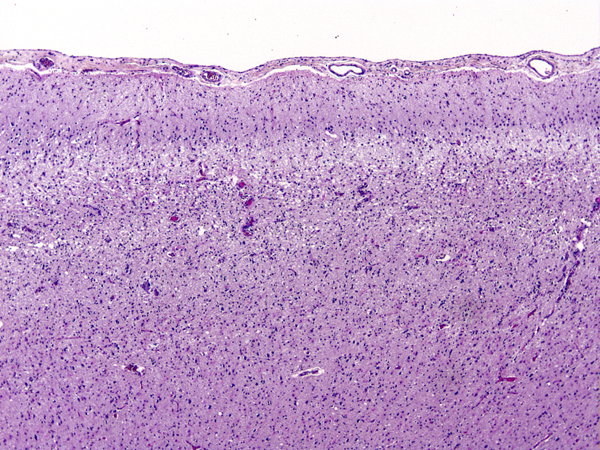 Pseudolaminar necrosis |
Damage of the cortex sometimes causes a band-like lesion, called pseudolaminar necrosis. There is also a regional variation in susceptibility to HIE. The cerebral cortex and striatum are more sensitive than the thalamus, and the thalamus in turn is more sensitive than the brainstem. The spinal cord may remain uninjured even when all the rest of the CNS is severely damaged. The most likely explanation for this selective vulnerability is that susceptible neurons produce more glutamate. In severe cases of HIE, not only neurons but glial cells are damaged as well. The key factor that converts selective neuronal necrosis to total tissue necrosis is probably lactic acidosis.
 Hippocampal sclerosis, left |
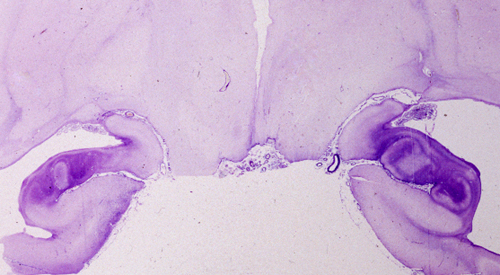 Hippocampal sclerosis |
The term HIE encompasses hypoxia, ischemia, hypoglycemia, and the efects of prolonged seizures. Shock and cardiac arrest develop at some point in most severe HIE cases, and the neuropathology is the same regardless of the initial event. Only rarely it is possible to identify the cause of HIE based on the pathological findings. For instance, bilateral hippocampal neuronal loss and gliosis (hippocampal sclerosis) without other lesions is seen in some patients with epilepsy or following cardiac arrest of short duration. Sparing of Purkinje cells suggests hypoglycemic encephalopathy but is also seen in HIE without hypoglycemia. In addition to causing hypoxia, CO binds to iron-rich neurons of the globus pallidus and substantia nigra, damaging these nuclei selectively.
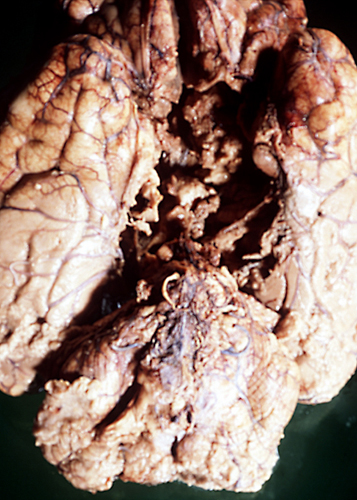 "Respirator brain" |
 Non-perfused brain |
Severe and protracted HIE, e.g. from cardiac arrest, damages the cortex, deep nuclei, and brainstem, and causes cerebral edema, resulting in brain death. Brain death may also result from primary brain pathology such as subarachnoid and intracerebral hemorrhage, traumatic brain injury, massive ischemic infarcts and other processes. (see below).
The common denominator in all these cases is increased intracranial pressure which exceeds mean arterial pressure, causing collapse of cerebral vessels and circulatory arrest. If such a patient is put on the respirator, the brain (under normal body temperature) undergoes an enzymatic autodigestion which may end in liquefaction. The term "respirator brain" that has been applied in such cases is misleading because the autolysis is not caused by the respirator. The term "non-perfused brain" is more accurate. Because circulation is arrested and all metabolic activity ceases, the non-perfused brain does not show any reactive changes (inflammation, macrophages, gliosis), only autolysis. Imaging reveals hypodensity due to edema and disintegration of brain tissue without enhancement.
In some instances, global ischemia causes bilateral, symmetric cerebral infarcts in the border zones between major arterial territories. Rarely, HIE involves the white matter, causing myelin damage and loss. Unravelling of damaged myelin results in vacuolization and a spongy appearance of the white matter in tissue sections. White matter damage is common in CO poisoning but may occur in other forms of HIE.
THE CLINICAL CONSEQUENCES OF HYPOXIC-ISCHEMIC ENCEPHALOPATHY
The most common pattern of injury in HIE is selective loss of sensitive neurons (pyramidal cells of CA1 of the hippocampus, layers 3, 5, and 6 of the neocortex, Purkinje cells, and striatal neurons). Mild HIE, such as a brief cardiac arrest, may affect CA1 pyramidal neurons of the hippocampus only. Bilateral hippocampal damage causes Korsakoff's amnesia. This is a memory disorder characterized by inability to retain new information (anterograde amnesia) and a less severe defect of recall of old memories (retrograde amnesia). Hippocampal amnesia (Korsakoff's amnesia) affects more severely episodic memory and less so semantic memory (see below)
MEMORY
PROCEDURAL: Learning skills (learning
how), e.g., how to write with the left hand,
if right -handed.
DECLARATIVE: two types
SEMANTIC: Memory for facts
(learning that), e.g., Kabul is the capital of
Afghanistan. EPISODIC: Memory for personally
experienced events, e.g., remembering where you
parked your car this morning.
Diffuse cortical, thalamic,
or combined neuronal loss (with intact brainstem)
results in dementia or the persistent vegetative
state (loss of cognitive functions and emotion
with preservation of sleep-wake cycles, autonomic
function, and breathing). The
medical, legal, and ethical issues revolving around
the persistent vegetative state were dramatized
in 2005 by the case of Terri
Schiavo.
More protracted
ischemic insults, which damage also the brainstem,
cause brain
death, a terminal clinical state characterized
by loss
of cerebral and brainstem function.
The clinical manifestations of brain death are complete
unresponsiveness, absence of brain stem reflexes, apnea,
electrical silence (flat EEG), and absence of cerebral
perfusion. The latter is probably due to blockage
of capillaries from endothelial swelling and cerebral
edema. Brain death can be distinguished clinicallly
from the persistent vegetative state and other conditions
that cause severe brain damage and coma. In most
cases, brain death leads to loss of vital functions.
Therefore, for legal purposes, brain death is the
equivalent of somatic or cardiorespiratory death.
THE WHITE MATTER IN HIE
Gray matter uses 2.5 times more ATP than white matter, and, for this reason, HIE damages primarily the cerebral cortex and deep nuclei. The white matter is only rarely affected. One exception is CO poisoning, in which the white matter is affected, along with but sometimes out of proportion to the pathology in the cortex and basal ganglia. In addition, the white matter, especially in subcortical regions, is involved diffusely in chronic ischemia, leading to a condition called leukoaraiosis. Leukoaraiosis (rarefaction of the white matter) is a neuroradiology term describing the loss of density of the white matter on CT and increased signal on T2 or FLAIR MRI. These findings can be explained by loss of myelin and axons and increased interstitial fluid. Leukoaraiosis is probably due to the chronic effect of microvascular disease associated with hypertension, diabetes, and angiopathies (cerebral amyloid angiopathy, CADASIL, Collagen 4A1 mutation-see page 2 and page 3 further on). Some of these angiopathies also cause intracerebral hemorrhage. Its most common substrate is small vessel disease. Patients with leukoaraiosis frequently also have lacunar infarcts. The extent of white matter pathology can be best appreciated by MRI, especially with newer techniques such as diffusion MRI, which measures the density of axons traversing the white matter. Pathological examination reveals loss of myelin and axons, vacuolation of the white matter and expansion of perivascular spaces, a finding known in classical neuropathology as état criblé (a sieve-like state). Binswanger encephalopathy, a white matter degeneration that has been linked to hypertension, is a pathological counterpart of some cases of leukoaraiosis. The pathogenesis of leukoaraiosis is unknown and probably has to do with nonspecific axon and myelin damage due to chronic ischemia. In that sense it may be considered as being analogous to periventricular leukomalacia (PVL), however, the term PVL should be restricted to the neonatal condition. Leukoaraiosis and the conditions that cause it are associated with cognitive decline and dementia, instability and gait abnormality with frequent falls, and bladder dysfunction.See also Small Vessel Disease
Further Reading
- Dirnagl U, Iadecola C, Moskowitz MA. Pathobiology of ischaemic stroke: an integrated view. Trends Neurosci. 1999;22:391-7. PubMed
- MacDonald JF, Xiong ZG, Jackson MF. Paradox of Ca2+ signaling, cell death and stroke. Trends Neurosci. 2006;29:75-81. PubMed
- O'Sullivan M. Leukoaraiosis. Pract Neurol 2008;8:26-38 PubMed
- Peters A, Schweiger U, Pellerin L, et al. The selfish brain: competition for energy resources. Neurosci Biobehav Rev. 2004;28:143-80.PubMed
Updated: January, 2022