HEMORRHAGIC STROKES (INTRACEREBRAL AND SUBARACHNOID HEMORRHAGE)
Approximately 15% to 20% of strokes are due to rupture of blood vessels causing intracerebral (parenchymal) or subarachnoid hemorrhage. The major causes of hemorrhagic stroke are hypertension, anticoagulants and bleeding disorders, cerebral amyloid angiopathy, ruptured arterial aneurysms, and arteriovenous malformations and other vascular anomalies. Intracerebral and subarachnoid hemorrhage are also very common in head trauma.
HYPERTENSIVE INTRACEREBRAL HEMORRHAGE
 Hypertensive basal ganglionic hemorrhage |
 Hypertensive pontine hemorrage. |
This hemorrhage results from rupture of small, penetrating arteries. Hypertensive angiopathy (small vessel disease) stiffens vessel walls and makes them fragile. This, in conjunction with increased pressure from within the lumen, causes vascular rupture and hemorrhage. Hypertensive hemorrhage is parenchymal and its most frequent sites are the basal ganglia and thalamus. Less commonly, it involves the cerebellum, the pons, and occasionally the subcortical white matter. Parenchymal hemorrhage disrupts brain tissue and accumulates rapidly within a few hours until pressure from the hematoma collapses the bleeding vessels. The blood clot is surrounded by a zone of compressed ischemic tissue. Vascular leakage, in this zone, causes cerebral edema, which increases over a few days. Thus, the hemorrhage causes focal neurological deficits and, more important, increased intracranial pressure. Improved control of hypertension in the last 20 years has led to a dramatic reduction in the incidence of hypertensive intracerebral hemorrhage.
HYPERTENSIVE ENCEPHALOPATHY
 Hypertensive encephalopathy. H&E |
 Hypertensive encephalopathy. PTAH |
Hypertensive encephalopathy (HE) is a syndrome characterized by severe headache, nausea and vomiting, papilledema, visual disturbances, seizures, confusion, and in severe cases coma. These clinical findings are seen in the background of severe (malignant) hypertension and are accompanied by retinopathy and nephropathy. In adults, HE is usually the culmination of severe chronic hypertension. In women with eclampsia and in children with new onset hypertension, it develops rapidly and with lower levels of blood pressure. Neuropathological studies of HE, done in the pre-MRI era, reveal fibrinoid necrosis and thrombosis of arterioles and capillaries and microinfarcts and microhemorrhages secondary to the vascular lesions. These changes are present thoughout the brain but are more prominent in the brainstem, especially the basis pontis. Initially, the vascular lesions were attributed to vasospasm. Current thinking favors loss of autoregulation and forcefull overdistention of blood vessels, leading to fluid extravasation and necrosis.
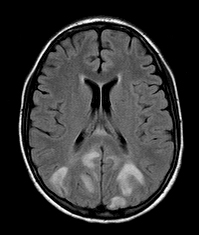 PRES |
The posterior reversible encephalopathy syndrome (PRES) is a recently described entity associated with severe hypertension or abrupt elevation of blood pressure, e.g. eclampsia, and administration of immunosuppressive and chemotherapeutic agents. It presents with headache, encephalopathy, seizures, and visual loss, and is thought to be caused by vascular dysregulation due to hypertension or endothelial dysfunction and damage of the blood-brain barrier caused by toxic agents. The MRI in PRES shows vasogenic edema most prominent in the occipital lobes (hence “posterior encephalopathy). In some cases the abnormality is more extensive, involving the thalamus, cerebellum and brainstem. The neuropathology of PRES is largely unknown. A recent autopsy report revealed microvascular changes including fibrinoid necrosis, indicating that the underlying mechanism of PRES is similar to the previously studied HE cases.
ARTERIAL ANEURYSMS
 Berry aneurysm |
 Large aneurysm at the cerebello-pontine angle |
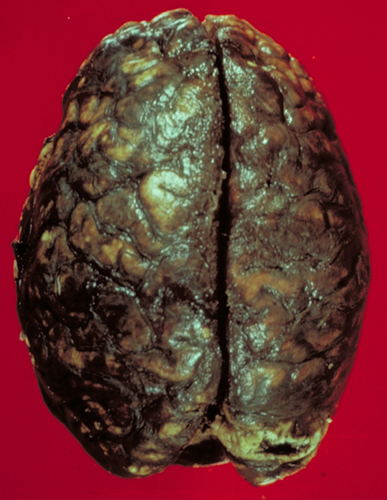 Subarachnoid hemorrage. |
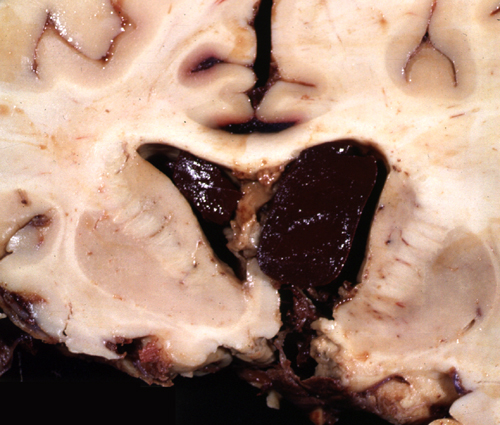 Intraventricular hemorrhage |
Intracranial aneurysms (IA), also referred to as saccular or berry aneurysms, develop in the walls of major cerebral arteries at branching points, where there are gaps in the media and internal elastica. The majority of them are on the circle of Willis and the first bifurcation of the middle cerebral artery. They are multiple in 20% of the cases. Nonruptured aneurysms are seen in 2% of adult autopsies. The defects in the vessel wall are present since birth but aneurysms are rare in children; they develop later in adulthood, due to gradual weakening of vessels from the constant force of even normal blood pressure and structural changes that occur with advancing age. They are more common in women than men and occur with increased frequency in patients with coarctation of the aorta, polycystic kidney disease, and connective tissue disorders. Risk factors for aneurysm rupture include hypertension, smoking, and alcohol abuse.
Clinical observations have established a familial incidence of IAs. A small proportion are inherited as an autosomal dominant trait and are linked to several genes and chromosomal loci, including some that encode collagen and other structural proteins that are found in vessel walls. There is an increased risk in first degree relatives of patients with aneurysms.
Large IAs can cause symptoms by compressing cranial nerves, vessels, and brain tissue, but their most feared complication is rupture. The vessels bearing the aneurysms are in the subarachnoid space. Consequently, their rupture causes subarachnoid hemorrhage (SAH). Blood spurts out of the ruptured aneurysm with a force that can tear the soft brain. If the stream of blood is directed toward the brain, it may cause intracerebral and intraventricular hemorrhage. The larger the aneurysm, the higher is the likelihood of rupture.
Typically, SAH from a ruptured IA causes a sudden severe headache with relative preservation of consciousness and without focal neurological deficits. Approximately one week later, vascular spasm develops, causing additional ischemia. Vasospasm affects arteries that are surrounded by subarachnoid blood clots and is triggered by products released from hemolyzed RBCs. A massive aneurysmal bleed raises intracranial pressure, resulting in arrest of cerebral perfusion, unconsciousness, and HIE. Hydrocephalus may develop due to blockage of CSF flow by subarachnoid clots and from meningeal fibrosis which results from their organization. About half of patients with aneurysmal bleeds die in six months, some from the first and most from recurrent bleeds. Survivors have serious long-term disabilities and a significant risk of rebleeding.
Fusiform aneurysms are vascular dilatations due to atherosclerosis. They are seen most commonly in the basilar artery and are associated with thrombosis and brainstem infarction and less frequently with rupture and subarachnoid hemorrhage.
ARTERIOVENOUS MALFORMATIONS (AVMs)
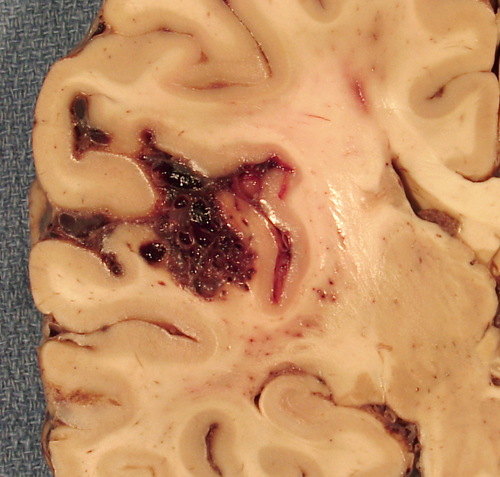 Arteriovenous malformation |
 Arteriovenous malformation |
AVMs are developmental abnormalities of cerebral vessels. They consist of a tangle of abnormal vessels interposed between a feeding artery and a draining vein. Most AVMs are in the distribution of the middle cerebral artery but they may occur anywhere. In addition to classical AVMs, there are several other related types of vascular anomalies and hamartomas that have similar manifestations. The abnormal vessels may be in brain tissue, in the subarachnoid space, or both. AVMs and other vascular anomalies cause seizures and neurologic deficits due to chronic compression and ischemia of brain tissue. Their most feared outcome is intracerebral and subarachnoid hemorrhage. There may be multiple episodes of bleeding over many years (sometimes since childhood) manifested by headaches, a single catastrophic bleed, or both. Patients with AVMs also have an increased incidence of aneurysms.
 CCM-MRI |
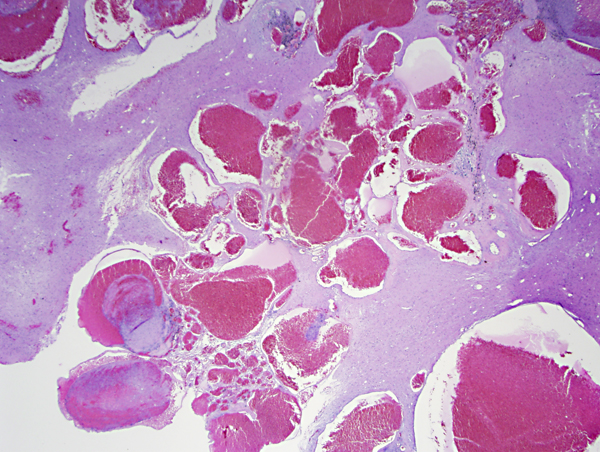 CCM-cavernoma |
A related vascular lesion, cerebral cavernous malformation (CCM) or cavernoma, consists of clusters of thin-walled cavernous vessels without intervening neural tissue. CCMs are present in 0.5% of the population. Approximately 15% of CCMs are familial, autosomal dominant, and the rest are sporadic. Rarely, CCMs develop following radiation, especially if that is given in childhood. Familial CCMs tend to be multiple. Up to half of patients with CCMs, more in some studies, remain asymptomatic through their lives. The rest have seizures, focal neurologic deficits, headaches, and cerebral bleeds, which are not as severe as those from AVMs. Symptoms usually develop between the second to fifth decade of life. Familial CCMs are caused by autosomal dominant mutations of 3 genes, KRIT1 (CCM1), CCM2 (OSM or malcavernin) or PDCD10 (CCM3). Patients with familial CCM have a germline mutation of one allele and acquire a somatic mutation of the other allele (second hit) during their lives. Patients with sporadic CCM have de novo (somatic) mutations. CCMs can be located in any part of the CNS. Twenty percent are located in the brainstem. The vessels of CCMs can range from tiny to a few centimeters. They have an incomplete endothelial lining without dense junctions and lack elastic or smooth muscle. They are clustered together without intervening neural tissue. Venous angioma or developmental venous anomaly (DVA), a frequent incidental finding on contrast-enhanced MRI, consists of radially arranged veins draining into a collector vein in an arrangement that has been likened to the head of Medusa. DVAs are usually asymptomatic and only rarely cause hemorrhage or neurological deficits. They may coexist with AVMs and CCMs, and the bleeding may be caused by these other lesions. Distortion of blood vessels due to chronicity and the effects of bleeding makes it difficult to classify these various vascular anomalies histologically.
STURGE-WEBER SYNDROME
 Sturge-Weber syndrome |
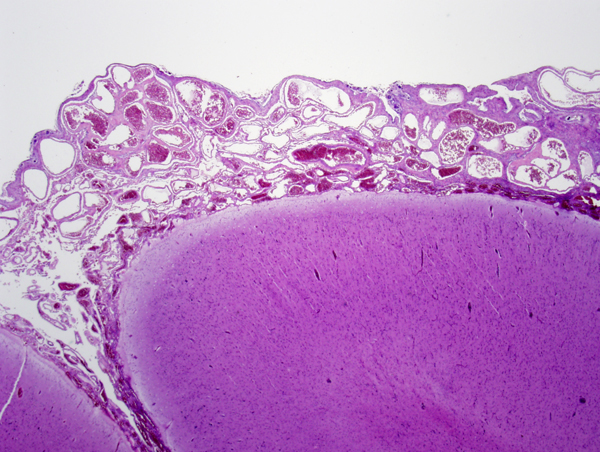 Sturge-Weber syndrome |
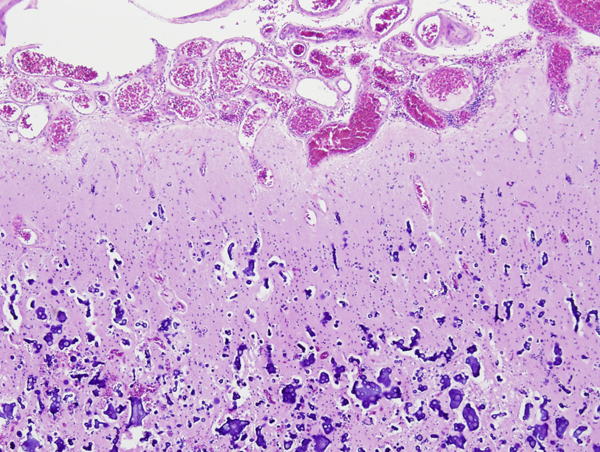 Sturge-Weber syndrome |
Sturge-Weber syndrome (SWS) is a rare sporadic neurocutaneous syndrome characterized by meningeal and ipsilateral cutaneous angiomatosis. The latter causes a port wine stain in the face in the distribution of the ophthalmic division of the trigeminal nerve. The key component of SWS is the meningeal angiomatosis. The majority of persons with facial angiomatosis do not have a meningeal component. The pathology in the brain consists of proliferation of blood vessels in the subarachnoid space, most frequently over the parietal and occipital lobe. The underlying brain shows calcified vessels in the cortex and white matter, neuronal loss, gliosis, and laminar cortical necrosis. The vascular malformation is probably static but, over time, stasis, venous congestion, and ischemia resulting from it, progressively damage the underlying brain. Patients with SWS have seizures, developmental delay, transient ischemic attacks, hemiparesis, headaches, and glaucoma.
CEREBRAL AMYLOID ANGIOPATHY
 Cerebral amyloid angiopathy |
 Lobar hemorrhage |
Cerebral amyloid angiopathy (CAA) is a frequent cause of parenchymal brain hemorrhage. Insoluble 8-10nm-thick amyloid fibrils are deposited in the walls of leptomeningeal and cortical small arteries, arterioles and capillaries. Similar to small vessel disease, this process destroys normal vascular elements, makes vessels fragile, causes thickening, and impairs their permeability. This pathology causes ischemic and hemorrhagic lesions. The ischemic lesions include microinfarcts and a diffuse ischemic degeneration of the white matter (leukoencephalopathy), which in imaging studies presents as leukoaraiosis (literally thinning of the subcortical and periventricular white matter). A similar entity, Binswanger encephalopathy, occurs in hypertension. The ischemic lesions of CAA cause dementia. The hemorrhagic lesions are microbleeds and large hemorrhages. Both these have a lobar distribution, i.e., they occur in locations other than the thalamus-basal ganglia, which are common sites of hypertensive bleeds. However, CAA-related hemorrhages can occur anywhere; a spontaneous cerebral hemorrhage in an elderly person without an apparent cause should raise suspicion for CAA. Occasionally, amyloid deposition incites a granulomatous angiitis.
Most CAA cases are due to deposition of betbeta amyloid (Aβ), the same peptide that is deposited in the plaques of Alzheimer's disease (AD). The majority of these are sporadic and a few are familial, autosomal dominant. The latter are a component of familial AD that is caused by trisomy 21 and mutations of the Amyloid Precursor Protein (APP), Presenilin, and other genes. CAA is present in the vast majority of patients with AD and contributes to their neurological deterioration. The risk factors for AD apply also to CAA. Moreover, the ApoE4 allele, which confers a high risk for AD, is also associated with lobar microbleeds, suggesting that the latter are a marker for CAA.About one third of persons older than 60 years also have CAA. A transient increase of vascular amyloid is seen in the course of immunization with Aβ42. Other familial CAAs are caused by mutations of Cystatin, Gelsolin, Transthyretin, and other genes. These patients do not have AD and the vascular amyloid has a different chemical composition.
OTHER CAUSES OF HEMORRHAGIC STROKE
A frequent cause of parenchymal brain hemorrhage is anticoagulant therapy. The incidence of anticoagulant-associated intracerebral hemorrhage has increased markedly in recent years, paralleling the increasing use of warfarin. Less frequently, intracerebral and subarachnoid hemorrhage is caused by cerebral angiitis (polyarteritis nodosa, granulomatous arteritis, SLE, bacterial and fungal arteritis) and other causes.
Further Reading
- Nahed BV, Bydon M, Ozturk AK, et al. Genetics of intracranial aneurysms. Neurosurgery. 2007;60:213-25. PubMed
- Flaherty ML, Kissela B, Woo D, et al. The increasing incidence of anticoagulant-associated intracerebral hemorrhage. Neurology 2007;68:116-21. PubMed
- Revesz T, Holton JL, Tammaryn L et al. Genetics and molecular pathogenesis of sporadic and hereditary cerebral amyloid angiopathies. Acta Neuropathol 2009;118:115-30 PubMed
- Rammos SK, Maina R, Lanzino G. Developmental venous anomalies: current concepts and implications for management. Neurosurgery 2009;65:20-9. PubMed
- Chester EM, Agamanolis DP, Banker BQ, Victor M. Hypertensive encephalopathy: a clinicopathologic study of 20 cases. Neurology 1978;28:928-39.PubMed
- Kheir JN, Lawlor MW, Ahn ES, et al. Neuropathology of a fatal case of Posterior Reversible Encephalopahty Syndrome. Ped Dev Pathol 2010 Feb 16 �Epub. PubMed
- Bartynski WS. Posterior reversible encephalopathy syndrome, part 1. Fundamental imaging and clinical features. AJNR Am J Neuroradiol 2008;29:1036-42. PubMed
Updated: June, 2023