The American Cancer Society estimates that 1,898,160 new cases of cancer will develop in 2021 resulting in 608,570 deaths. Compared to this, the number of estimated new primary brain tumors (BT), 24,530, or 1.26%, is relatively small. However, if one adds metastases from other cancers to the brain, the number of BT increases significantly. Primary BT are projected to claim 18,600 lives in 2021, 2.9% of all cancer deaths. The incidence of BT shows a peak in childhood followed by a decline till 25 years. After this, there is a continuous increase with advancing age. Cancer is second only to accidents as a cause of death in children and adolescents, 1 to 19 years old. It causes 10.5% of all deaths. In that age group, BT, with an average annual age-adjusted incidence rate of 5.42 per 100,000, are the most frequent neoplasm, accounting for about 18% of all cancers. The second most common cancer in that age group is leukemia. The CNS is among the three leading sites of cancer mortality in the first three decades of life. The most frequent BT (all ages) is meningioma (35.6%) followed by glioblastoma (15.4%).
HISTOGENESIS - CLASSIFICATION OF BRAIN TUMORS
As in all cancers, the tumor cells of BT display some features of normal brain cells. This similarity may be obvious in the microscopic appearance and fine structure of tumor cells (especially in low-grade tumors) or revealed by their marker expression patterns and is the basis of the classification of BT (see table below). This phenotypic-histologic classification has been in use for many decades and is also a histogenetic classification, i.e., it reflects our notions of the presumed cells of origin of BT. In the latter part of the 20th century, histological and immunohistochemical studies refined the phenotype and identified many new types of BT. By the end of the century, brain tumors were classified essentially on the basis of their morphology. An abbreviated classification is given below.
CLASSIFICATION OF BRAIN TUMORS
| TUMORS OF
NEUROGLIAL CELLS Gliomas Diffuse astrocytoma Anaplastic astrocytoma Glioblastoma Oligodendroglioma Diffuse midline glioma Pilocytic astrocytoma Pleomorphic xanthoastrocytoma Subependyal giant cell astrocytoma Other gliomas Neuronal Tumors Dysembryoblastic neuroepithelial tumor Ganglioglioma Gangliocytoma Central neurocytoma Tumors of ependymal cells Ependymoma and variants Choroid plexus tumors Choroid plexus papilloma and carcinoma EMBRYONAL TUMORS Medulloblastoma Atypical rhabdoid/teratoid tumor PINEAL TUMORS Pinealoma and pineoblastoma |
|
TUMORS OF CRANIAL
AND
SPINAL NERVES
Schwannoma Neurofibroma Malignant peripheral nerve sheath tumor MENINGEAL TUMORS Meningioma MESENCHYMAL TUMORS Hemangioblastoma Hemangiopericytoma/solitary fibrous tumor Other soft tissue tumors Chondro-osseous tumors CEREBRAL LYMPHOMAS GERM CELL TUMORS Germinoma Embryonal carcinoma Endodermal sinus (yolk sac) tumor Teratoma TUMORS OF THE SELLAR REGION Craniopharyngioma METASTATIC TUMORS |
The term "glioma" refers to all glial tumors in general but is also used instead of astrocytoma.
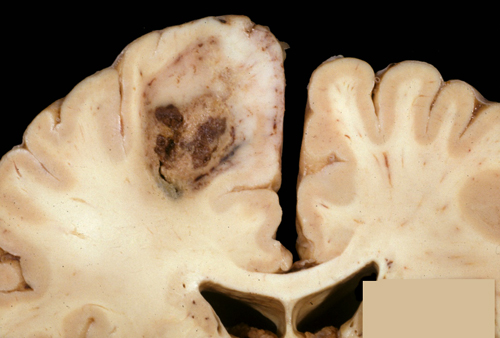 Intra-axial tumor |
 Extra-axial tumor |
The terms "intra-axial" and "extra-axial," used in radiological descriptions, mean "in brain or spinal cord tissue" and "extrinsic to brain" respectively. For instance, astrocytoma and oligodendroglioma are intra-axial; meningioma and Schwannoma are extra-axial. The term anaplasia describes the cellular atypia and loss of differentiation that are associated with malignant tumors.
In adults, metastatic BT outnumber primary ones but are less frequently subjected to biopsy or studied at autopsy. Among primary BT, the most common ones are meningioma, glioblastoma, and astrocytoma. In children, primary BT are more common than metastatic, and the most frequent among them are pilocytic astrocytoma and medulloblastoma. The most common tumors of the spinal cord are Schwannoma, meningioma, and ependymoma. The majority of BT in children arise in the cerebellum and brainstem (infratentorial). Most BT in adults arise in the cerebrum (supratentorial).
MOLECULAR-GENETIC ASPECTS OF BRAIN TUMORS
The study of BT was greatly enhanced with the introduction of molecular methods during the 21st century. Now, the molecular changes of BT are an integral part of their diagnosis. The 2021 WHO classification of BT more than 100 entities and identifies 42 distinctive molecular profiles of BT and it is likely that this will continue to expand. These molecular profiles correlate with the histological phenotype and clinical behavior and have significant influence on treatment options. The upshot of these developments is that BT are histologically and genetically diverse (and some may be unique) and this diversity is reflected in the current classification and clinical approach to BT.
It had been assumed, until recently, that gliomas arise by transformation of normal glial cells. These cells were thought to be the only cells that had the capacity to divide, even though there is little evidence that they do so in the mature brain. This view is now being re-examined in the light of recent discoveries about neural stem cells (NSC), the multipotential precursors that give rise to neurons and glial cells. Until recently, NSCs were thought to be present mainly during fetal life, but it is now obvious that they also exist during post-natal life. They are more numerous and active during childhood, when the brain continues to develop, but are also found in the mature brain, especially around the ventricles, in the hippocampus, and in other locations.
It is likely that gliomas and other tumors of neuroglial cells arise from NSCs. NSCs are capable of proliferation and divergent differentiation. The genes that are expressed in these BT, including Nestin, EGFR, PTEN, Hedgehog, and others (see also below) are the same ones that are involved in neurogenesis and gliogenesis. This suggests that aberrant activation of developmental genetic programs in NSCs gives rise to BT. Such activation results in emergence of transformed cells with an enhanced ability to proliferate and migrate. The NSC origin explains the higher incidence of gliomas and medulloblastoma in children, the fact that some BT are composed of immature cells, the inclusion of neurons in some BT, and the presence of multiple cell types within the same tumor, such as oligoastrocytoma. The ultimate expression of multipotentiality is the development, in the brain, of teratomas, i.e., tumors containing derivatives of all 3 germ layers, similar to tumors arising in the gonads.
Some of the genes that are involved in neoplasia-oncogenes- promote cell growth and others –tumor suppressor genes-have the opposite action. Oncogenes code for growth factors, growth factor receptors, cytoplasmic signal transduction molecules, and nuclear transcription factors. When these proteins are inappropriately or excessively expressed (due to gene amplification, translocation, mutation, and other mechanisms), cells change their phenotypes and gain functions (such as moving through the extracellular matrix, inducing angiogenesis) that enhance their ability to survive, compared to their normal neighbors.
The proteins of tumor suppressor genes, together with other catalytic and inhibitory factors, regulate the cell cycle and restrain cell proliferation. Loss of both copies of tumor suppressor genes (from mutation, chromosomal deletion, aberrant methylation, and other mechanisms) leads to unrestrained cell proliferation. Such mutation or deletion may result from acquired (environmental) damage of one allele, then the other. Individuals who have one defective allele of a tumor suppressor gene in their germline tend to develop tumors at a young age, and this trait is transmitted to their children. The best known tumor suppressor genes are the retinoblastoma (Rb) gene on chromosome 13q, and p53 gene on 17p. Both these tumor suppressor genes are involved in the pathogenesis of BT.
The genetic changes that are involved in BT are briefly outlined in the following descriptions of each individual BT. The mainstay in the diagnosis and classification of BT is still their microscopic phenotype. However, molecular alterations are progressively taken into account and provide information that is crucial for diagnosis, prognosis, and, more important, for management of BT. Information about molecular changes can be obtained by various genetic testing methods and immunohistochemistry.
BT are initially derived from a single progenitor, but later, the genetic instability that is associated with rapid cell replication causes molecular and chromosomal changes to snowball, generating multiple tumor cell clones. This adds to the phenotypic heterogeneity and is reflected in the karyotypes and gene expression patterns of BT.
OTHER FACTORS IMPLICATED IN THE PATHOGENESIS OF BRAIN TUMORS
Age: Embryonal tumors of the brain and other organs (cerebellar medulloblastoma, adrenal neuroblastoma, occur predominantly in children. Neurogenesis and neuronal migration in the cerebrum are largely completed by midgestation, but in the cerebellum they continue for the first year of life. Production of glial cells is very active in childhood. The brisk cell division that is associated with these processes gives the chance for new gene defects to emerge and for inherited ones to be unmasked. The cerebellum is the most cellular part of the CNS (granular neurons outnumber all other neurons in the brain together) and takes the longest to develop. It is no coincidence that it is the most frequent site of BT in children.
Radiation: An increased incidence of BT, especially meningiomas, has been reported in patients who have received radiation to the head (even low-dose radiation) for a variety of reasons. Children with ALL who have been treated with craniospinal radiation are at high risk for developing meningiomas and gliomas. These tumors emerge many years after radiation has been given and appear to be on the rise.
Chemical Carcinogens: A variety of substances can induce mesenchymal and glial CNS tumors in animals by direct intracerebral inoculation and by oral and parenteral administration. The most potent neurocarcinogens in experimental animals are nitroso compounds (NOCs). NOCs are present in foods (cured meats, fish, vegetables), cosmetics, rubber products, even in beer and water, and are also synthesized in the mouth, stomach, and bladder by nitrosation of amines and amides in the diet. Given their ubiquitous nature, it is likely that they are also involved in human BT.
Immunosuppression: Cerebral lymphoma, usually B-cell, is frequent in patients with AIDS, renal transplants, congenital immunodeficiency syndromes, and other immunosupressed states. The finding of EBV DNA suggests that some of these tumors arise from EBV-infected B-cells.
DIAGNOSIS OF BRAIN TUMORS
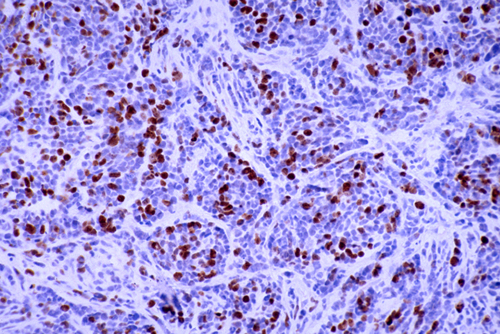 Ki67 immunostain |
The laboratory evaluation of BT entails morphology morphology (the study of their microscopic, immunohistochemial and ultrastructural features) supplemented by molecular findings, when the latter are available. Molecular and chromosomal analysis play an important role for diagnosis and for designing a biological approach to treatment. Immunohistochemistry can now be used to reveal molecular changes underlying BT. Genetic testing can be performed on paraffin-embedded tissue but it is a good practice to set aside frozen tissue for molecular testing, if adequate samples are available.
The clinical behavior of BT is expressed by their grade, which correlates with their prognosis. The 2021 WHO grading of BT includes 4 grades, with grade 1 being the least malignant and grade 4 the most malignant. Some BT such as diffuse astrocytoma evolve from low-grade to high-grade while others, such as medulloblastoma are high grade from the start. Grading is based largely on histological features (cellularity, cellular and nuclear atypia [anaplasia], proliferative index and necrosis). The simplest proliferative index is a count of mitoses. More elaborate evaluations use the Mib-1 antibody raised against the Ki-67 antigen which is expressed during all phases of the cell cycle except the G0 phase. Information about the labeling index (LI) of BT has been published and it is a useful prognostic measure.
A layered diagnosis of BT is currently in use. This includes an overall combined statement of tissue and molecular findings, the histological diagnosis, WHO grade, and a separate line of molecular information.
Further Reading
- Louis DN, Perry A, Wesseling P, et al. The 2021 WHO Classification of Tumors of the Central Nervous System: a summary Neuro-Oncology 2021;23:1231-1251. PubMed
- Crino PB. mTOR: A pathogenic signaling pathway in developmental brain malformations. Trends Mol Med 2011;17: 734-42. PubMed
- Aronica E, Becker AJ, Spreafico R. Malformations of Cortical Development. Brain Pathol 2012;22:380–401. PubMed
- Ostrom QT, Gittleman H, Liao P,et al. CBTRUS Statistical Report: Primary Brain and Central Nervous System Tumors Diagnosed in the United States in 2007–2011. Neuro-Oncology 2014;16:iv1–iv63.PubMed
- Cotter JA. An update on the central nervous system manifestations of tuberous sclerosis complex. Acta Neuropathologica 2020; 139:613–624.PubMed
Updated:November, 2021