MEDULLOBLASTOMA AND OTHER EMBRYONAL TUMORS
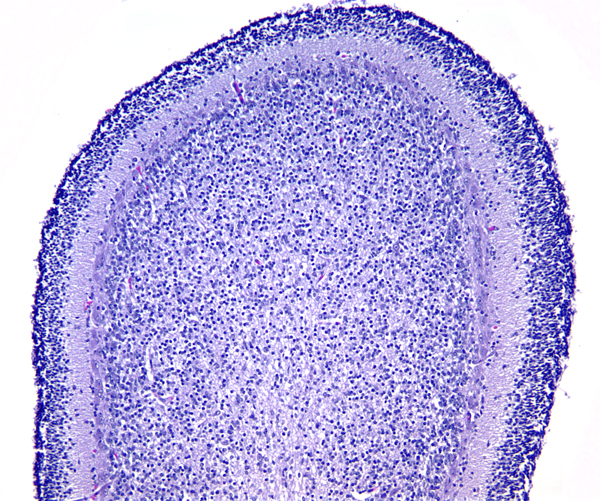 The external granular layer |
In some statistics, medulloblastoma is listed as the most common BT in children, representing 20% of all BTs. In other accounts, including the author's practice, it is the second most frequent BT in children after pilocytic astrocytoma. Most medulloblastomas occur in the first decade of life. There is a second peak in the early 20s. Several genetic tumor syndromes, including the Turcot (familial adenomatous polyposis) and Gorlin (nevoid basal cell carcinoma) syndrome are associated with medulloblastoma. Medulloblastoma is an embryonal tumor of the brain, analogous to Wilms tumor of the kidney and neuroblastoma of the adrenal. Its embryonal nature is underlined by its high incidence in infants and children and by its undifferentiated, immature appearance, which resembles developing neural tissue. Some medulloblastomas are thought to arise from stem cells located in the subependymal matrix and the external granular layer (EGL) of the cerebellum. This layer is formed from precursor cells that migrate from the rhombic lip (the most lateral and dorsal part of the hindbrain) to the surface of the developing cerebellum where they divide and differentiate. Neurons then move inwards forming the permanent granular layer of the cerebellar cortex. The EGL persists until the beginning of the second year of life. Different stem cells from the subependymal matrix around the fourth ventricle give rise to the cerebellar nuclei and Purkinje cells. Some medulloblastomas (WNT-activated see below) are thought to arise from the dorsal brainstem.
 Medulloblastoma |
 Medulloblastoma |
 Medulloblastoma |
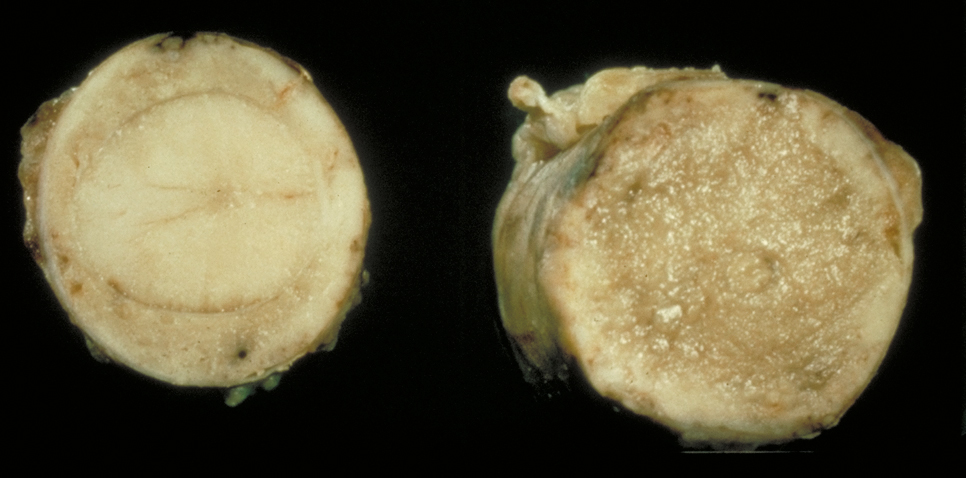
Medulloblastomas are tumors of the cerebellum, arising more frequently in the midline, especially in the posterior vermis, adjacent to the roof of the fourth ventricle. A few of them arise in the cerebellar hemispheres (see table below). On MRI imaging, they are mostly compact, isointense, and show contrast enhancing. On gross examination, they are soft, pink-red, and well demarcated. They can block the fourth ventricle and the aqueduct, causing hydrocephalus. They are rapidly growing tumors and present over a period of weeks or a few months with signs and symptoms of increased intracranial pressure and cerebellar dysfunction.
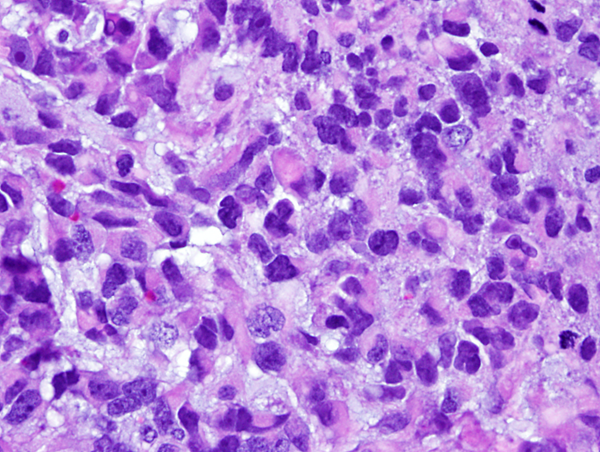
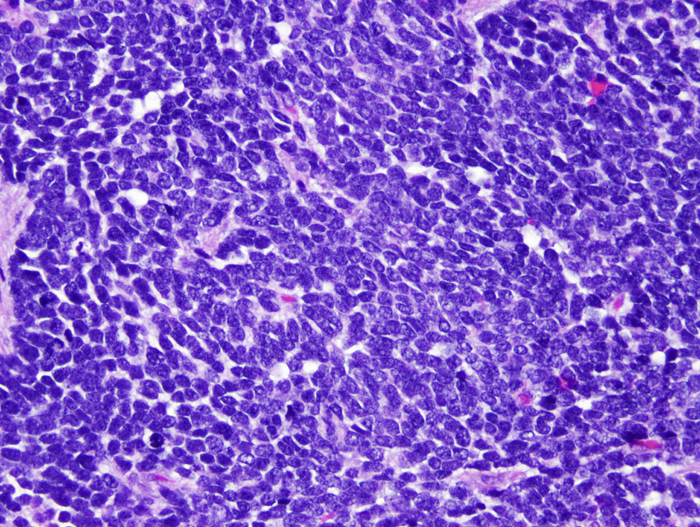 Classic Medulloblastoma |
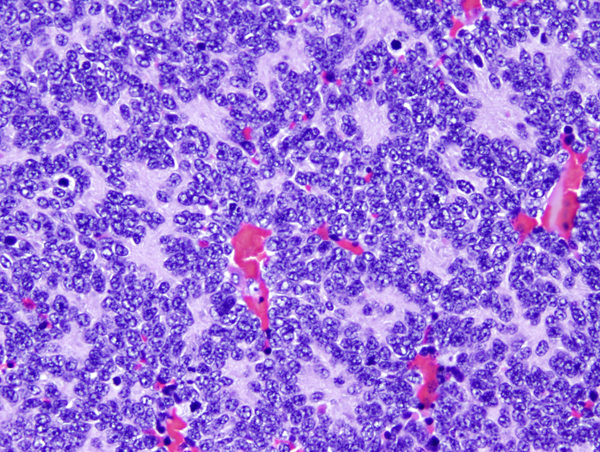 Medulloblastoma: Homer-Wright rosettes |
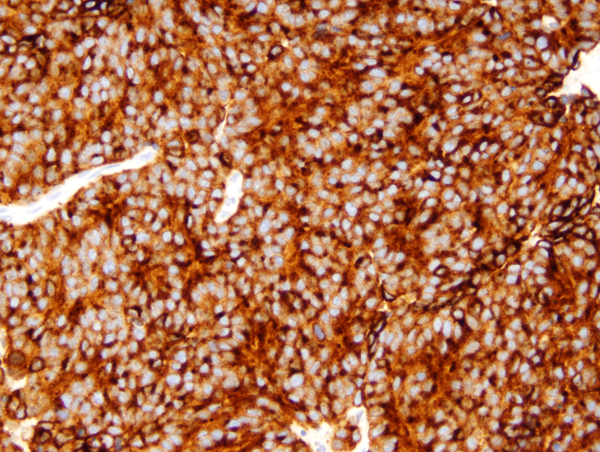 Medulloblastoma: Synaptophysin |
Four histological varians of medulloblatoma are recognized: classic, desmoplastic/nodular, large cell-anaplastic, and medulloblastoma with extensive nodularity.
Classic medulloblastoma (the majority) is a highly cellular tumor composed of diffuse masses of small, undifferentiated oval or round cells. Some medulloblastomas show neuronal, glial and other differentiation. Neuronal differentiation is manifested by neuropil and rosette formation. Rosettes are groups of tumor cells arranged in a circle around a fibrillary center. Mature neurons may also be found infrequently. Glial differentiation in some tumors is reflected by GFAP-positive cells. There may also be differentiation along oligodendroglial or ependymal lines. More unusual lines of differentiation result in formation of striated muscle cells (medullomyoblastoma) and melanin-producing cells.
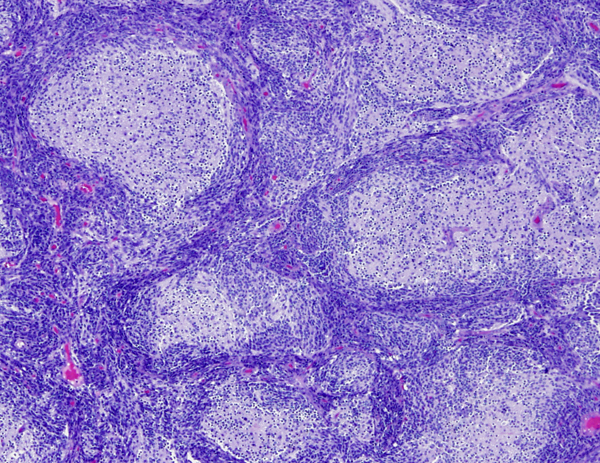 Desmoplastic/nodular medulloblastoma |
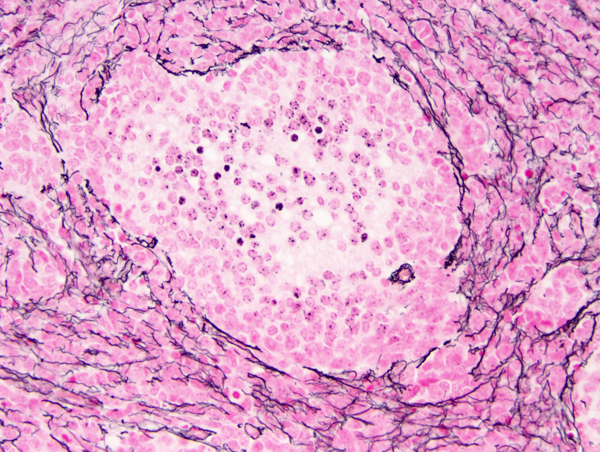 Desmoplastic/nodular medulloblastoma |
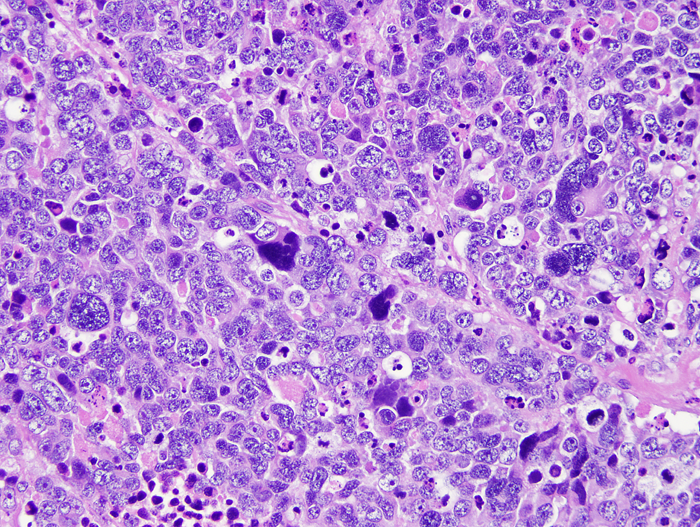 Large cell/anaplastic medulloblastoma. |
Desmoplasmic/nodular (D/N) medulloblastoma is called nodular because of its architecture and desmoplastic because it is permeated by fine collagen (reticulin) fibers that give it a firm consistency. The centers of the nodules (pale islands) are reticulin-free. They are less cellular than the surrounding densely packed small cells and are composed of larger cells with neurocytic differentiation. Desmoplasia may occur when a superficially located tumor encroaches upon the arachnoid membrane. This is a reactive change and, if there are no nodules, it is not equated to D/N medulloblastoma. D/N medulloblastoma is more common in infants and may have a better prognosis than the classic form.
Medulloblastoma with extensive nodularity may be difficult to distinguish from D/N. It is characterized by expanded nodules with neurocytic differentiation . It occurs also in infants and has a good prognosis.
Large-cell/anaplastic medulloblastoma (LCA) shows large, anaplastic nuclei with a high rate of mitosis and apoptosis. This variant has poor prognosis.
Distinct molecular signatures correspond to some of these clinicopathological phenotypes (see table below).
The genesis of MB is driven by genetic pathways that are also involved in the development of the cerebellum. Abnormalities in these pathways convert stem cells to tumor cells. Three molecular groups of medulloblastoma have been described: WNT-activated, SHH-activated(subdivided into PTP53-mutant and TP53-wild-type), and non-WNT/non-SHH. The WNT and SHH groups have been defined in greater detail. The third group, non-WNT/non-SHH, which includes the majority of medulloblastomas, is less well characterized and is further subdivided into two subsets, Group 3 and Group 4. These molecular subtypes and groups are not homogeneous in their clinical and pathological phenotypes, and their molecular characterization is in progress. Their main features are partially listed in the table below. They can be identified by molecular analysis and surrogate immunohistochemical markers. In the future, more precise molecular characterization may lead to personalized therapy for these tumors.
| MOLECULAR PATHWAY | WNT | SHH | Group 3 | Group 4 |
|---|---|---|---|---|
| Genes involved | Beta-Catenin mutation, monosomy 6 |
PTCH1 deletion, SUFU deletion, MYCN amplification | MYCN |
MYCN amplification Isochromosome 17q |
| Clinical profile | Older children and adults, good prognosis | Infants, children and adults, good to intermediate prognosis | Infants and children, poor prognosis | Older children and adults, the most common form, intermediate prognosis |
| Tumor location | IV ventricle, infiltration of dorsal brainstem | Cerebellar hemispheres | Midline, filling the 4th ventricle | Midline, filling the 4th ventricle |
| Histology | Classic | D/N, LCA | Classic, LCA | Classic, LCA |
| Cell of origin | Precursors around the IV ventricle | EGL | EGL | Unknown |
| Tumor syndrome | Turcot | Gorlin | None | None |
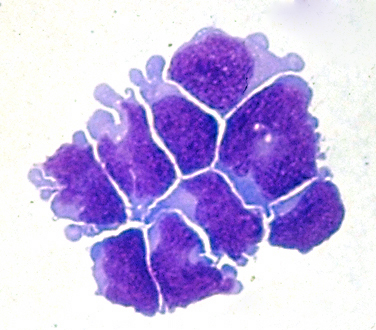 Medulloblastoma in CSF |
 Medulloblastoma growing around the spinal cord |
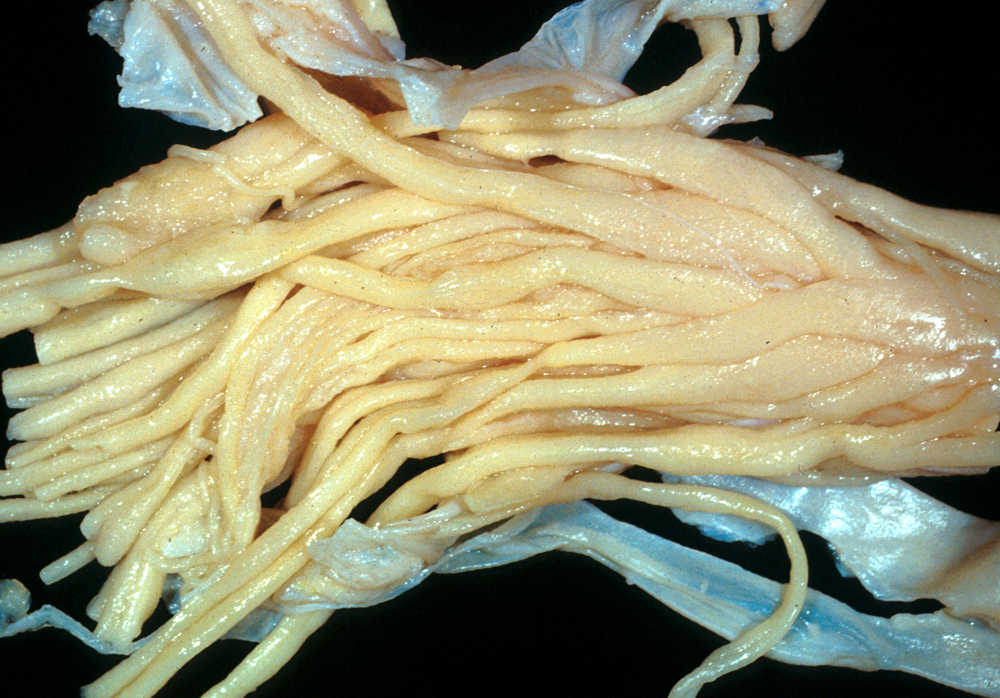 Medulloblastoma infiltrating the roots of the cauda equina. |
Although there are significant differences in prognosis depending on histological and molecular profile, medulloblastoma is classified as a grade IV tumor. It infiltrates and destroys brain tissue and tends to seed the subarachnoid space and spread along the walls of the ventricles. Leptomeningeal dissemination occurs more frequently in medulloblastoma than any other BT. The CSF shows high protein and low glucose, and contains tumor cells. CSF cytology is used to monitor the spread of the tumor. Extracranial metastases occur rarely, usually after operation or shunting. Treatment combines resection, to reduce the tumor mass and decompress the fourth ventricle, shunting of the lateral ventricles, radiation of the tumor bed and the entire neuraxis, and intrathecal chemotherapy.
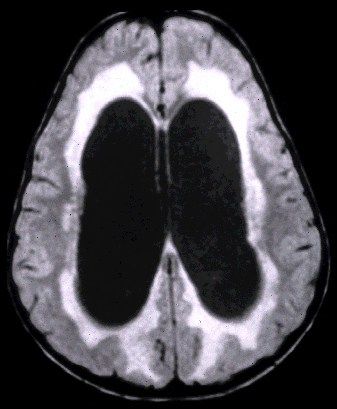 Hydrocephalus in medulloblastoma |
Medulloblastoma, cerebellar pilocytic astrocytoma, and other posterior fossa tumors compress the aqueduct and fourth ventricle (or grow in these spaces) causing hydrocephalus. They usually present with symptoms of increased intracranial pressure such as as morning headache, vomiting, and blurred vision. Fundoscopic examination reveals papilledema. Other symptoms include ataxia, strabismus, nystagmus, and stiff neck. The latter is a sign that the tumor is extending through the foramen magnum. Absence of focal deficits compounded by the difficulty of getting a history from a young child may lead to the wrong diagnosis, such as gastroenteritis or aseptic meningitis. A lumbar puncture, in this setting, can induce cerebellar tonsillar herniation ending up in disaster.
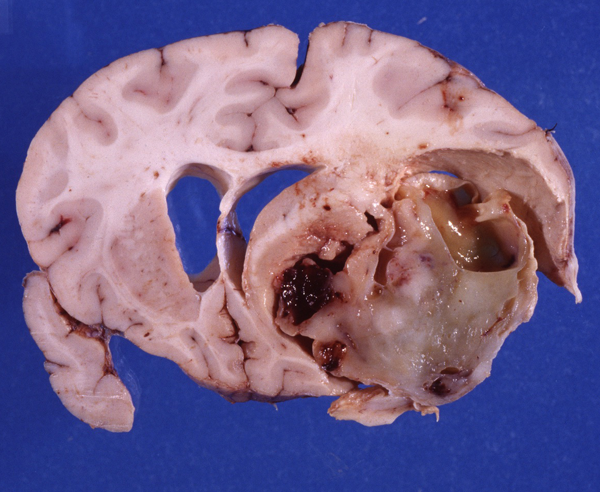 PNET Malignant supratentorial embryonal tumor. Note midline shift and subfalcial herniation. Image provided by Dr Maie Herrick. |
Other embryonal tumors are less frequent. Tumors identical to medulloblastoma that arise in the cerebral hemispheres are called supratentorial primitive neuroectodermal tumors (PNETs). Some cerebellar and extracerebellar embryonal tumors resemble closely adrenal neuroblastoma. These tumors are called cerebral neuroblastomas.
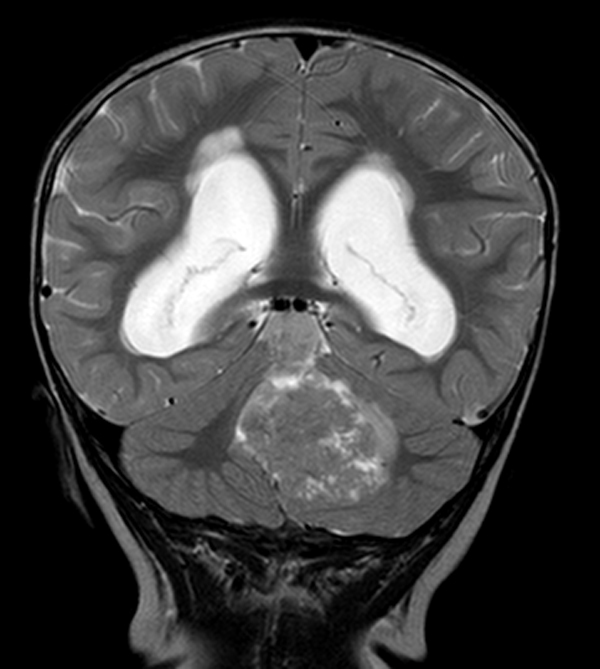 ATRT MRI |
 ATRT A |
 ATRT-INI1 |
 ATRT-FISH |
One important embryonal trumor is Atypical Teratoid Rhabdoid Tumor, (ATRT), a highly aggressive neoplasm affecting very young children. ATRT arises in the cerebellum and extracerebellar locations and is composed of rhabdoid cells and diverse other neuroectodermel and mesenchymal elements, hence the term teratoid. It has a distinct molecular signature shared by extraneural rhabdoid tumors, i.e., loss of both copies of the INI1 gene, located on 22q11.3. The product of this gene is involved in chromatin remodeling. On immunohistochemical staining, INI1 is absent in tumor nuclei and present in non-neoplastic nuclei. Loss of one copy of INI1 in the germline results in the Rhabdoid Tumor Predisposition Syndrome, in which patients develop renal and extrarenal rhabdoid tumors, including ATRTs, and choroid plexus tumors.
Further Reading
- Gilbertson RJ, Ellison DW. The Origins of Medulloblastoma Subtypes. Annu Rev Pathol Mech Dis 2008;3:341-65. PubMed
- Dhall G. Medulloblastoma. J Child Neurol 2009;24:1418-30. PubMed
- Northcott PA, Korshunov A, Pfister SM and Taylor MD. The clinical implications of medulloblastoma subgroups. Nat Rev Neurol 2012; 8:340–351. PubMed
- Millard NE, De Braganca KC. Medulloblastoma. J Child Neurol 2015;J Child Neurol. 2015 Sep 2. pii: 0883073815600866. [Epub ahead of print]. PubMed
- Orr B A. Pathology, diagnostics, and classification of medulloblastomaBrain Pathol. 2020; 30: 664–678. PubMed
Updated: February, 2022