TUMORS OF GLIAL CELLLS-B: OTHER GLIOMAS
PILOCYTIC ASTROCYTOMA-WHO GRADE I
Pilocytic astrocytoma (PA) is a biologically and histologically distinct form of astrocytoma of children and young adults and is the most frequent BT in children. Most PAs arise in the cerebellum, hypothalamus, and optic chiasm. Some arise in the cerebral hemispheres and other locations.
 PA of the cerebellum |
 PA of the pons |
 Hypothalamic PA |
Grossly, pilocytic astrocytomas are circumscribed and often cystic with a solid mural nodule. Histologically, they are sparsely cellular tumors without anaplasia or mitoses. They have a biphasic pattern, consisting of cellular and fibrillary perivascular areas, alternating with loose microcystic zones. Some PAs are mostly compact, consisting usually of fiber-like spindle cells.
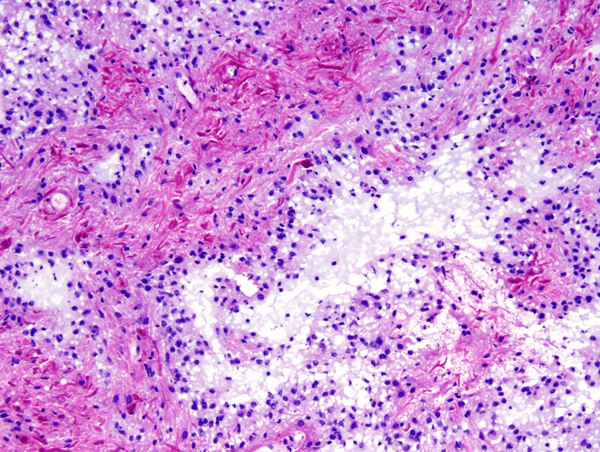 Pilocytic astrocytoma |
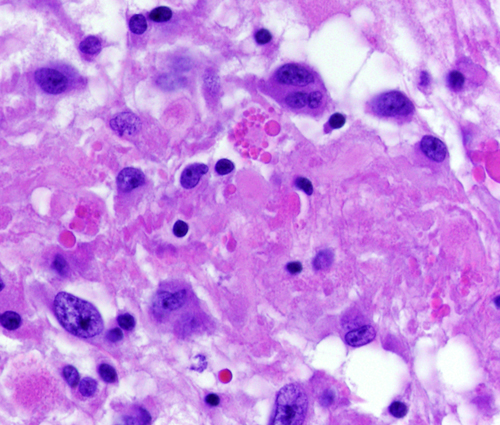 Eosinophilic granular bodies |
 Pilocytic astrocytoma |
The tumor cells often contain Rosenthal fibers and eosinophilic granular bodies. The word pilocytic (hair cell) refers to the fiber-like appearance of the tumor cells and their fibrillary stroma, but large parts of these tumors, especially in the loose areas, do not fit this description. Some PAs contain clear (oligodendroglioma-like) cells, multinucleated cells, and calcifications. PAs are highly vascular and enhance with contrast injection. Most PAs are biologically low grade and do not evolve into more malignant tumors. Surgical excision of cerebellar PA (even partial resection in some instances) sometimes results in permanent cure.
The vast majority of sporadic cerebellar and midline PAs and a smaller proportion of extracerebellar ones show activation of BRAF. This is most commonly caused by fusion of BRAF with KIAA1549, a contiguous gene on 7q34, producing a fusion gene that lacks the BRAF regulatory domain. KIAA1549:BRAF duplication/fusion activates the MAPK oncogenic signaling cascade, which is involved in the pathogenesis of PA. This BRAF alteration is not seen in PA associated with NF1. The same pathway can be activated by the V600E BRAF mutation, which is common in pleomorphic xanthoastrocytoma, ganglioglioma, and, less frequently, in other low-grade pediatric gliomas. The V600E mutation is also seen in colorectal cancer, melanoma, and thyroid carcinoma. These BRAF changes can be detected by molecular analysis. In addition to their diagnostic and prognostic value, these findings open the road for specific pharmacological interventions aiming to inhibit the activated pathways.
PLEOMORPHIC XANTHOASTROCYTOMA-WHO GRADE II
 PXA |
 PXA |
 PXA |
 PXA |
 PXA |
 PXA |
Pleomorphic xanthoastrocytoma (PXA) is a rare glioma of children and young adults. Typically, it is a superficially located, supratentorial, intra-axial cystic tumor that has a solid mural nodule. It has the tendency to invade the subarachnoid space, and, rarely, it arises in the meninges, thus mimicking the neuroimaging appearance of meningioma. The "xantho" part of the term refers to the presence of vacuolated tumor cells containing lipid droplets. The "pleomorphic" designation indicates that the tumor displays significant cellular and nuclear pleomorphism. In spite of this, PXA does not have a high proliferative index. Most PXAs are WHO grade II but may undergo malignant transformation (WHO grade III), especially when they recur after resection. Eosinophilic granular bodies are a frequent histological marker of PXA. A network of fine collagen (reticulin) fibers permeates PXA, and lymphocytic inflammation is frequently present. Most PXAs carry the V600E BRAF mutation.
DIFFUSE MIDLINE GLIOMA-WHO GRADE IV
 Diffuse midline glioma |
 Diffuse midline glioma |
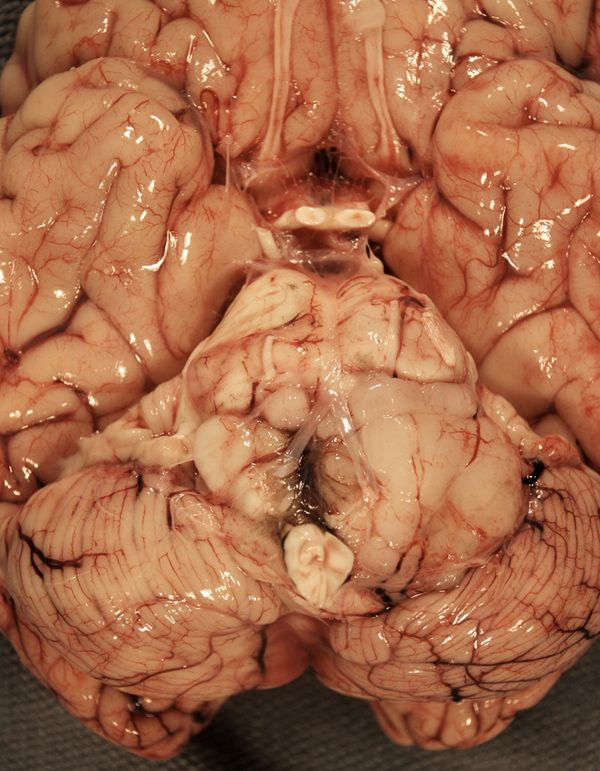 Diffuse midline glioma |
Diffuse midline glioma (DMG) is a WHO grade IV glioma that affects children. It is a diffusely infiltrating astrocytic neoplasm that arises in the pons, thalamus, and other midline structures. Some of these tumors are also called “Diffuse Intrinsic Pontine Glioma” (DIPG). Pontine and medullary DMG present insidiously with brainstem signs (cranial nerve deficits, long tract signs) and progress inexorably, usually causing death in 2 years. The diagnosis is usually made by MRI which shows a large area of T1-hypointense and T2-hyperintense signal involving the pons or thalamus extensively. Biopsy is done infrequently and may be difficult to interpret, not the least because of the small sample size dictated by the location of the tumor. At autopsy, most DMGs consist of poorly differentiated glial cells that infiltrate the involved parts diffusely, leaving neurons and white matter tracts intact. This explains the mild early symptoms compared to the size of the lesion. In some cases, more aggressive features identical to glioblastoma develop. The majority of DMGs have a distinctive molecular alteration, which is a mutation at position K27 of histone coding genes. The K27M H3 mutation can be detected by immunohistochemistry.
Further Reading
- Horbinski C. To BRAF or not to BRAF: Is That Even a Question Anymore? J Neuropathol Exp Neurol 2013;72:2-7. PubMed Huse JT, Rosenblum MK. The Emerging Molecular Foundations of Pediatric Brain Tumors. J Child Neurol 2015;30;1858-50. PubMed
- Rodriguez FJ, Lim KS, Bowers D, Eberhart CG. Pathological and Molecular Advances in Pediatric Low-Grade Glioma. Annu Rev Pathol Mech Dis 2013;8:361-79. PubMed
Updated: August, 2017