GENETIC TUMOR SYNDROMES
Listed below are certain familial disorders that are associated with an increased risk of CNS tumors. Together, these and other familial tumor syndromes account for a small proportion (between 1% and 4%) of BT.
| SYNDROME | GENE/LOCUS | NS TUMORS | OTHER TUMORS |
|---|---|---|---|
| NF1 | NF1/17q | Plexiform neurofibroma, MPNST, optic and other gliomas | Pheochromocytoma, GIST |
| NF2 | NF2/22q | Vestibular and PN shwannoma, meningioma, other brain tumors | NA |
| TS | TSC1/9q, TSC2/16p | SEGA | Renal AMLs, lung LAM, cardiac rhabdomyoma |
| VHL | VHL/3p | Hemangioblastoma | Renal cell carcinoma, pheochromocytoma |
| Li-Fraumeni | TP53/17p | Astrocytoma | Breast carcinoma, bone and soft tissue sarcoma |
| Turcot | FAP/5q | Medulloblastoma, GBM | GI polyps, colorectal cancer |
| Gorlin | PTCH/9q | Desmoplastic medulloblastoma | Nevoid basal cell carcinoma, Odontogenic keratocysts |
AML: Angiomyolipoma; GIST: Gastrointestinal stromal tumor; LAM: Lymphangioleiomyomatosis; MPNST: Malignant peripheral nerve sheath tumor

Von Recklinghausen neurofibromatosis (VRNF - Neurofibromatosis type 1-NF1), one of the most common genetic disorders, is autosomal dominant and is caused by mutations of a gene on chromosome 17q that encodes a protein called neurofibromin, which is involved in control of cell proliferation and acts as a tumor supressor. Patients with VRNF have a variety of tumors, including bilateral optic nerve astrocytomas, and plexiform neurofibromas and MPNSTs. VRNF also causes café au lait spots of the skin, axillary and inguinal freckles, dysplasia of the sphenoid wing and other skeletal abnormalities, fibromuscular dysplasia of arteries, and other lesions.
Bilateral acoustic neurofibromatosis (BANF - Neurofibromatosis type 2-NF2) is an autosomal dominant condition characterized by acoustic and spinal schwannomas, meningiomas, ependymomas and lenticular opacities. It is due to inactivation of the NF2 gene on chromosome 22q. This gene encodes a structural protein, schwannomin or merlin, which has tumor suppressor activity.
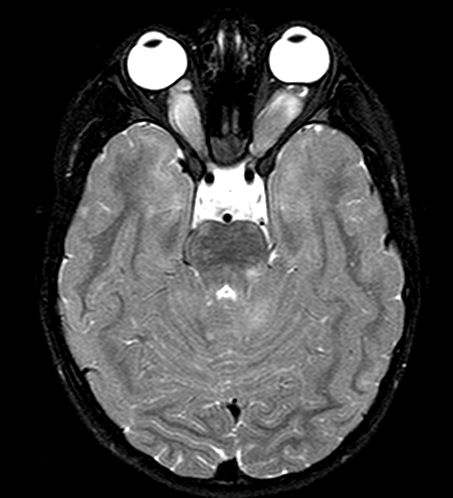
 Subependymal nodule |
 SEGA |
 SEGA |
 Cortical tubers |
Tuberous Sclerosis Complex (TSC) is an autosomal dominant condition that affects the brain, skin, kidneys, lungs, heart, and other organs. It is characterized by several major and minor features that are used as diagnostic criteria. Major features are cortical tubers, subependymal nodules (SENs), subependymal giant cell astrocytomas (SEGAs), hypomelanotic skin macules, shagreen patches, retinal nodular hamartomas, ungual or periungual fibromas, cardiac rhabdomyomas, pulmonary lymphangiomyomatosis, and renal angiomyolipomas. Minor features include dental pits, intraoral fibromas, multiple renal cysts, “confetti” (hypopigmented) skin lesions, nonrenal hamartomas, and retinal achromatic patches. The diagnosis of TSC can be established if 2 major or one major and 2 minor criteria are present. TSC patients have seizures, intellectual disability, ADHD, and other neuropsychiatric disorders.
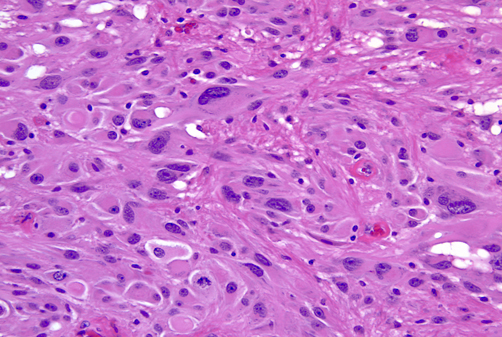
The cortical tubers are abnormal, broad (potato-like), firm gyri. They represent focal malformations of cortical development, similar to focal cortical dysplasia type IIB, and have an abnormal cytoarchitecture which includes large, often bizarre pyramidal cells and balloon cells that express neuronal and glial markers in a background of gliosis. These cells may be present in the white matter underlying the tubers. Tubers are epileptogenic. The SEGAs consist of variable size, often large or giant astrocytes and have frequent calcifications. Despite their cellular atypia, they are slowly growing tumors but can cause hydrocephalus because they are located at the foramina of Monro. The SENs are located most frequently at the angle between the thalamus and caudate nucleus and have a similar architecture to SEGAs
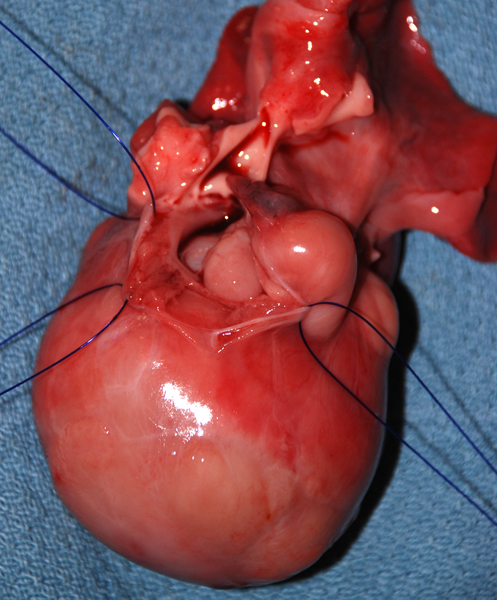 Cardiac rhabdomyoma |
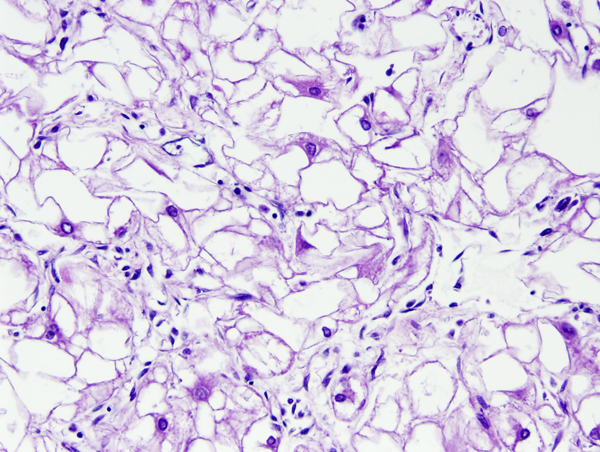 Cardiac rhabdomyoma |
One third of TSC cases are caused by mutations of the TSC1 gene on 9q34, which encodes hamartin, and two thirds are caused by mutations of TSC2 on 16p13.3, which encodes tuberin. Hamartin and tuberin form a functional complex which suppresses the activity of the mammalian Target Of Rapamycin (mTOR), a serine/threonine kinase that plays a central role in multiple processes involved in brain development, including neuronal proliferation, neuronal growth, and axon formation. Interestingly, the SEGAs and SENs are located in areas that, in the developing brain, correspond to the germinal matrix . Rapamycin is an antibiotic, antifungal and immunosuppressant that is also a potent antagonist of mTOR. Overactivation of mTOR signaling (caused by mutations of TSC1 and TSC2 and other regulatory proteins) leads to cortical malformations, including tubers, focal cortical dysplasia type IIB, ganglioglioma, and hemimegalencephaly. The latter is a mosaic disorder affecting one hemisphere. mTOR hyperactivation is evident in pathological specimens from these disorders, which have been grouped under the umbrella term "mTORopathy". The key features of mTORopathies are abnormal cortical cytoarchitecture, abnormal cells (dysplastic neurons and balloon cells), intractable seizures, and cognitive abnormalities. mTOR overactivation has also been demonstrated in NF1, Sturge-Weber syndrome, Cowden syndrome, PMSE (Polyhydramnios, Megalencephaly, Symptomatic Epilepsy), and Proteus syndrome. Treatment with mTOR inhibitors (Rapamycin, Everolimus) causes regression of SEGAs and other TSC lesions.
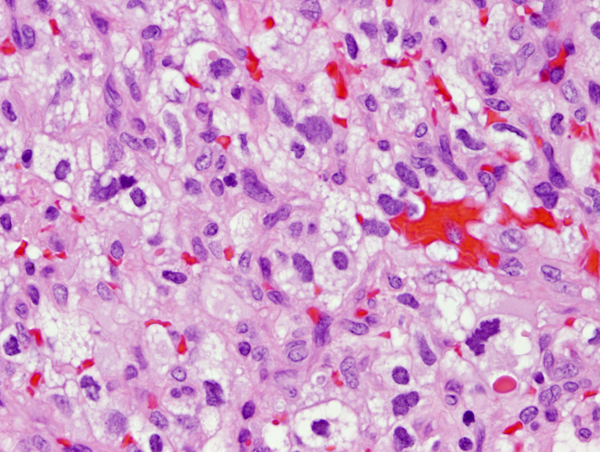 Cerebellar hemangioblastoma |
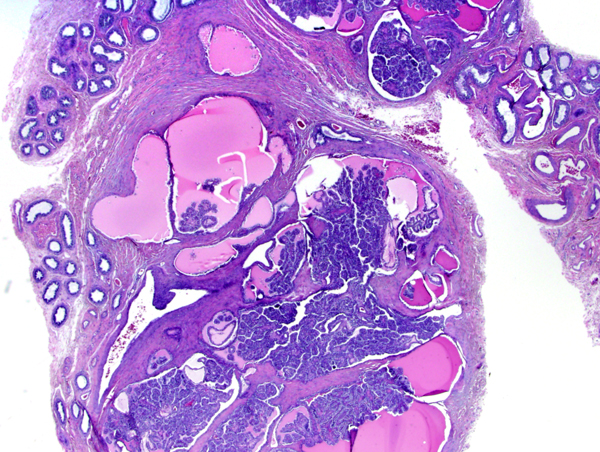 Papillary cystadenoma of the epididymis in VHL |
Von Von Hippel-Lindau (VHL) disease is an autosomal dominant disease, caused by mutations of the VHL tumor suppressor gene on chromosome 3p25.3. The product of this gene is involved in mRNA transcription and multiple other functions. VHL disease is associated with hemangioblastomas of the cerebellum, retina and spinal cord, cysts and clear cell carcinomas of the kidneys, pheochromocytomas, cysts of the liver and pancreas, pancreatic tumors, epididymis cysts, cystadenomas of the broad ligament and endolymphatic sac tumors of the middle ear. Hemangioblastoma of the cerebellum is usually a cystic lesion with a protruding nodule which is composed of capillaries and interspersed clear cells.
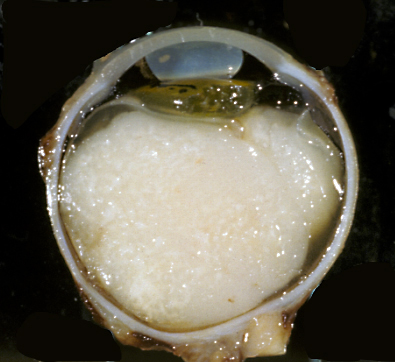 Retinoblastoma |
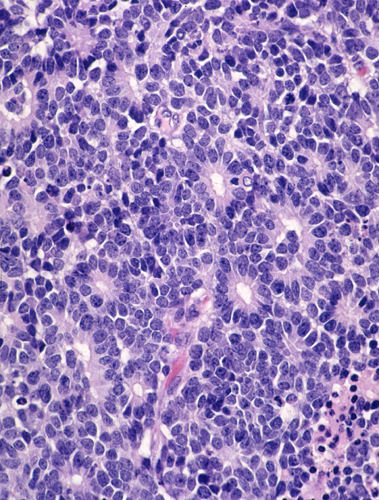 Retinoblastoma |
Retinoblastoma. Retinoblastoma is a malignant embryonal tumor of the eye similar to medulloblastoma. It is composed of “small blue cells”, which are frequently arranged in characteristic circular formations, the Flexner-Wintersteiner rosettes. Eighty five percent of retinoblastomas are sporadic. Fifteen percent are autosomal dominant and are often bilateral and multiple. Tumor development is due to deletion of the Rb tumor suppressor gene on chromosome 13q14. Familial tumors have a germline mutation resulting in one defective copy of the gene. Tumors develop when the remaining normal copy is altered due to a somatic mutation. The same principle is involved in the pathogenesis of other tumors.
Moved from page 1:March 2022