CONGENITAL MYOPATHIES
Congenital myopathies (CMs) are genetic muscle disorders characterized by structural abnormalities of myofibers and, some of them, by abnormal protein accumulation in the sarcoplasm. The nomenclature of CMs is based on their pathological changes. The most common CMs are core myopathies, nemaline myopathy, and centronuclear myopathy.
CMs have a wide clinical range. At the severe end of the spectrum are conditions that are apparent even before birth because of decreased fetal movements and polyhydramnios. Such cases cause severe hypotonia and weakness at birth, respiratory failure requiring ventilatory support and inability to swallow requiring feeding gastrostomy. About 12% of patients with CM die in the first year of life. Once patients get over the neonatal period, the disease is usually either static or slowly progressive and may be compatible with a normal life span. Patients often have proximal and facial weakness, dysmorphic facial features, kyphoscoliosis, and other physical problems, and lag behind peers in physical prowess. CK is normal, as there is no myonecrosis. The diagnosis is made by detecting the specific structural abnormality on a muscle biopsy using light and electron microscopy, and increasingly also by genetic testing. CMs are genetically diverse. Different gene mutations can cause the same clinicopathological phenotype and the same mutation can cause different clinicopathological phenotypes. The most common CMs are core myopathies, and the most frequent genetic change is RYR1 mutation. CMs are rare, but they are important because they cause neonatal hypotonia. The differential diagnosis of neonatal hypotonia includes also perinatal asphyxia, metabolic disorders, congenital CNS abnormalities, and spinal muscular atrophy.
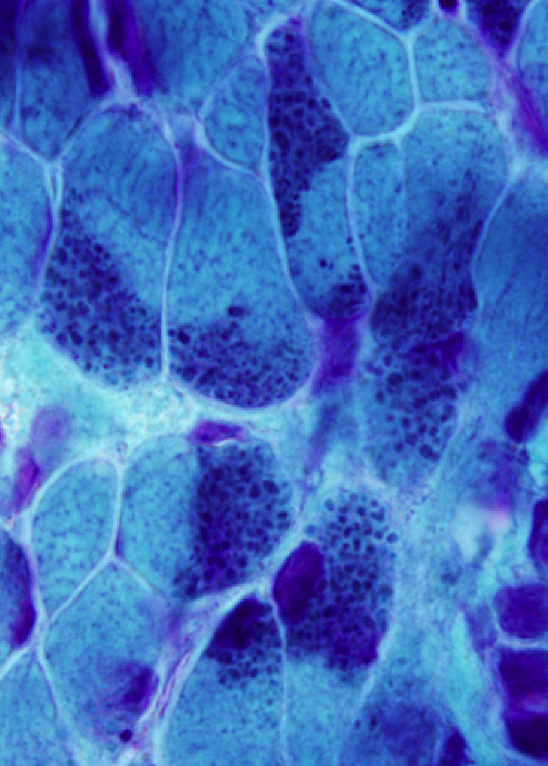 Nemaline myopathy |
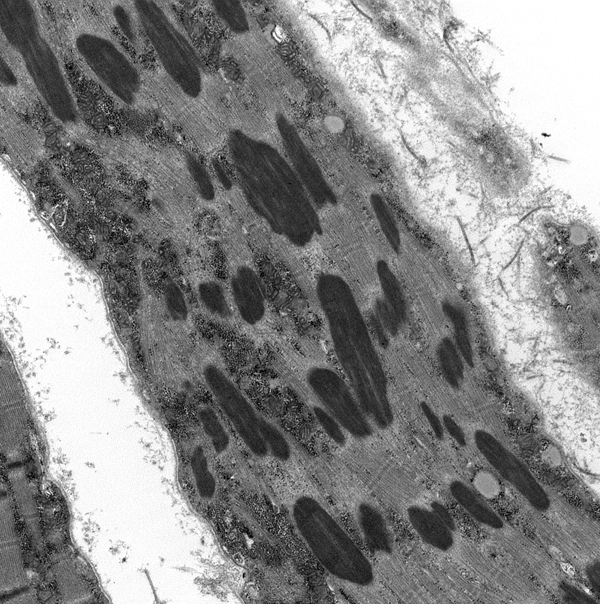 Nemaline myopathy |
Nemaline or rod body myopathy (NM) (Greek nema, thread) shows small and disorganized myofibers which contain rod-shaped structures composed of a-actinin, the main protein of Z-dics. The rods are located in the sarcoplasm and in some cases in the nuclei. They stain red or purple with the modified Gomori trichrome stain and have a lattice structure, similar to Z-discs, on EM. NM is caused most commonly by dominant mutations of ACTA1 (encoding skeletal a-actin) and recessive mutations of NEB (encoding nebulin) and less frequently by mutations of other genes. There are 6 clinical forms of NM ranging from the severe neonatal form, which presents at birth with severe hypotonia and may be fatal in infancy to the adult-onset variant that has a milder phenotype. Nemaline rods occur infrequently in other neuromuscular disorders, including dematomyositis and HIV myopathy.
Centronuclear myopathy |
 Congenital myotonic dystrophy |
Centronuclear (myotubular) myopathy is characterized by small myofibers with central nuclei, and central areas without contractile filaments -that contain aggregates of mitochondria and other organelles. The most frequent entity in this group is X-linked myotubular myopathy, caused by mutations of MTM1 on Xq28. This CM presents with severe hypotonia and weakness at birth or prenatally and may be fatal in infancy. Severe
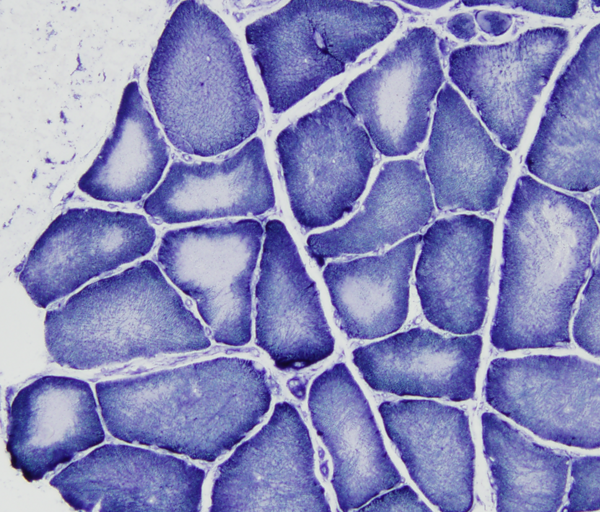 Central core disease |
 Central core disease |
In central core disease (CCD) myofibers have a central area (core) lacking oxidative enzyme activity. The cores consist of disorganized contractile filaments without mitochondria. A similar lesion, target fiber, occurs in denervation. Minicores and multi-minicores are variations of the same pathology. CCD is caused by autosomal dominant or recessive mutations of RYR1 which encodes the skeletal muscle ryanodine receptor, a calcium channel on the sarcoplasmic reticulum. Some CCD variants with multi-minicores are caused by mutations of SEPN1and several other genes. The product of SEPN1 interacts with the ryanodine receptor in regulating calcium homeostasis in muscle. Patients with CCD have hypotonia and weakness at birth or starting in infancy. Most patients have a normal lifespan but severe forms of CCD may be fatal in infancy. Some mutations of RYR1 (and CCD) confer susceptibility to malignant hyperthermia, a disorder of skeletal muscle calcium regulation.
 CFTD |
Congenital fiber type disproportion (CFTD). The CMs described above share in common a predominance and smallness of type 1 fibers. Some children with the clinical phenotype of CM have only type 1 fiber predominance and smallness without other structural abnormalities. These cases have been called CFTD. It is not clear if CFTD represents a disease entity or a “finding”. Genetic testing in CFTD cases reveals mutations of genes that are also associated with other CMs.
Further Reading
- Colombo I, Scoto M, Manzur AY, et al. Congenital myopathies. Natural history of a large pediatric cohort. Neurology 2015;84:28-35.PubMed
Updated: June, 2022