HUNTINGTON'S DISEASE
Huntington's Disease (HD) is a progressive and ultimately fatal, autosomal dominant neurodegenerative disorder that begins usually in the 4th to 5th decade of life and is characterized by behavioral changes, psychiatric abnormalities, involuntary movements including chorea, oculomotor abnormalities, diverse neurological (motor and sensory) defects, and dementia. Neurodegeneration characteristically affects the striatum resulting in loss medium size spiny, enkephalin-containing internuncial neurons in the caudate nucleus and putamen. It begins in the caudal portions of the caudate and putamen and advances rostrally. Biochemical analysis of the striatum shows loss of neurotransmitters, including GABA, acetylcholine and glutamate, which correlates with the loss of neurons. Decreased GABA and unbalanced dopamine activity in the striatum causes chorea. While the striatal pathology is the hallmark of HD, there is also severe neuronal loss involving the deeper layers of the cerebral cortex, thalamus, cerebellum and brainstem nuclei. Cortical neurodegeneration accounts for the personality changes, psychiatric abnormalities motor and sensory defects and dementia. Loss of corticostriatal connections may also contribute to or be the primary cause of chorea. Involvement of other areas accounts for the diverse neurological findings.
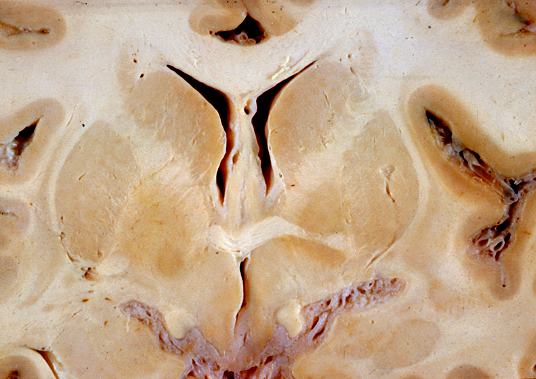 Normal caudate nuclei. |
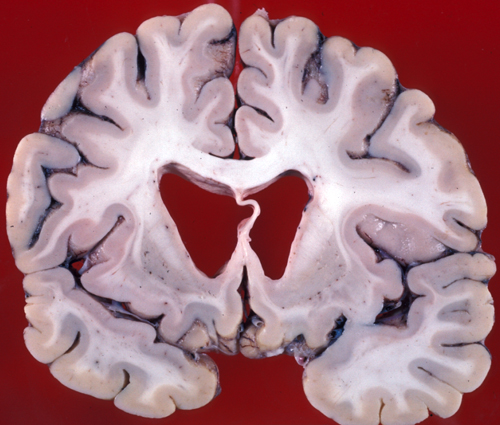 HD. Atrophy of the caudate nuclei. |
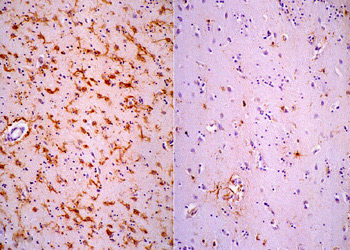 GFAP immunostain, showing gliosis. Left: HD; right: normal caudate nucleus. |
Gross examination of the brain reveals atrophy of the caudate nucleus and putamen, dilatation of the anterior horns of the lateral ventricles and cortical atrophy. The striatal pathology is obvious on MRI in advanced cases. Milder changes may be detected on neuroimaging years before the onset of symptoms. Microscopic examination shows shrunken (degenerating) neurons, ballooned neurons containing a basophilic substance, and axonal swellings and neuronal intranuclear inclusions containing ubiquitin. There is loss of neurons and proliferation of reactive astrocytes, which, in advanced cases, causes the affected nuclei to appear more cellular than normal.
The molecular abnormality of HD is CAG trinucleotide expansion of the HTT(HUNTINGTIN) gene on chromosome 4p. This adds a polyglutamine segment to huntingtin. This protein is widely expressed throughout the brain. It is thought that the CAG-expanded huntingtin has a toxic function. However, huntingtin is a key cellular protein involved in cellular transport and is important for cell viability. Therefore, loss of huntingtin function may also contribute to the pathogenesis of HD. The CAG triplet expansion is probably due to malfunction of cellular processes that repair strand breaks and remove mispaired bases from DNA. The expanded huntingtin, conjugated with ubiquitin, forms aggregates (inclusions) in the nuclei cytoplasm and dendrites of affected neurons. These inclusions can be detected by immunohistochemistry using antibodies to huntingtin. These findings suggest that there is an error in the proteolytic degradation of the expanded huntingtin. Normally, this degradation takes place in cellular chambers called proteasomes and involves conjugation of huntingtin with ubiquitin. Impairment of this process apparently causes huntingtin-ubiquitin complexes to be translocated into the nuclei.Healthy persons have 6-35 repeats. Adult-onset patients have 40-50 repeats. A high number of repeats is associated with early onset and more rapidly progressing disease. The expanded CAG repeat is unstable and may increase in size in successive generations. This causes the disease to appear at a younger age and more severe form, a phenomenon known as anticipation. Anticipation occurs more commonly if the mutated allele is inherited from the father. CAG repeats on other genes are also seen in spinobulbar muscular atrophy (Kennedy's disease), Machado-Joseph disease, an autosomal dominant ataxia seen mainly in Portugese of Azorean descent, and cerebellar degenerations.
A rare form of chorea, beginning in adolescence or early adult life, is associated with an erythrocyte abnormality (chorea with acanthocytosis). Pathologically, this entity is similar to Huntington's disease but is caused by a different gene defect. Sydenham Chorea is a transient disorder in rheumatic fever caused by antibodies that react with neuronal antigens in the basal ganglia.
Further Reading
- Martinez-Vicente M, Sovak G, Cuervo AM. Protein degradation and aging. Exp Gerontol. 2005;40:622-33. PubMed
- Rüb U, Seidel K, Heinsen H, et al. Huntington's disease (HD): the neuropathology of a multisystem neurodegenerative disorder of the human brain. Brain Pathol 2016;26(6):726-740. doi: 10.1111/bpa.12426.PubMed