ATAXIA AND CEREBELLAR DEGENERATION
Ataxia can be caused by lesions that interrupt the sensory input to the cerebellum (spinal or sensory ataxia), pathology of the cerebellar cortex resulting in incorrect execution of cortical signals (cerebellar ataxia), or by a combination of both (spinocerebellar ataxia). In terms of genetics, ataxias can be divided into 3 groups listed below.
| Friedreich's ataxia (FRDA)-an autosomal recessive ataxia caused by GAA repeats on the frataxin gene |
| Spinocerebellar ataxias(SCA)-a group of autosomal dominant ataxias (25 entities at last count), caused by CAG repeats on multiple chromosomal loci |
| Cerebellar ataxias-a diverse group of sporadic diseases that cause cerebellar degeneration and degeneration of other anatomical systems |
In addition to the inherited ataxias, cerebellar degeneration is caused by a variety of acquired conditions including prion disease, HIE, nutritional deficiency, and inherited metabolic diseases.
 Friedreich's ataxia |
Friedreich's ataxia (FRDA), the most frequent inherited ataxia, is an autosomal recessive, multisystem diseorder which begins usually before age 20 with gait ataxia, proprioceptive and superficial sensory loss, weakness and atrophy of the extremities, and spasticity with extensor plantar responses. Most patients die of cardiomyopathy. Foot deformity (pes cavus) and scoliosis are common, and there is also an increased incidence of blindness, deafness, and diabetes. FRDA is primarily a sensory neuropathy. Loss of sensory ganglion cells and degeneration of their axons in peripheral nerves, dorsal roots, and posterior columns deprives the cerebellum of sensory input that is necessary to coordinate movement. There is also transsynaptic loss of neurons in the dorsal nuclei of Clarke and degeneration of the dorsal spinocerebellar tracts. The cerebellar cortex is normal but there is loss of neurons in the dentate nuclei, the main source of cerebellar output, and degeneration of the superior cerebellar peduncles. There is loss of upper motor neurons (Betz cells) and degeneration of the lateral corticospinal tracts.
DNA analysis in FRDA shows GAA trinucleotide repeat expansion of the frataxin (FXN) gene on chromosome 9q21, which silences the gene. The product of this gene is a mitochondrial matrix protein which is involved in iron homeostasis. This suggests that FRDA is due to mitochondrial dysfunction and oxidative stress. FRDA may also result from mutations of one allele of the frataxin gene in association with GAA expansion of the other allele. Nutritional deficiency of the antioxidant vitamin E causes similar spinal cord lesions. A hereditary form of ataxia with vitamin E deficiency is caused by mutations of the alpha-tocopherol transfer protein.
Autosomal dominant spinocerebellar ataxias (ADSCAs). There is no other group of neurodegenerative diseases with the clinical and pathological diversity of the ADSCAs. This diversity is even more impressive considering that all these diseases have a common underlying molecular defect, namely CAG triplet expansion. If the expansion lies in a coding sequence, it is translated into a polyglutamine (polyQ) stretch of the affected protein. Similar to Huntington’s disease (which is also caused by CAG repeats), the ADSCAs show the phenomenon of anticipation, i.e. lengthening of the CAG repeat with earlier onset and more severe disease in successive generations. The expansion occurs more often with paternal transmission.
In addition to ataxia, the ADSCAs cause parkinsonism and other extrapyramidal manifestations, weakness and fasciculations, spasticity, ophthalmoplegia, retinal degeneration and optic atrophy, cognitive impairment, dementia, and peripheral neuropathy. The core neuropathology is cerebellar degeneration (in some cases OPCA-see below) with additional degenerations involving the basal ganglia, substantia nigra, motor neurons of the brainstem and spinal cord, the spinocerebellar and olivocerebellar tracts, cerebral cortex, and other systems. Microscopic examination sows loss of Purkinje cells and neurons in other affected nuclei. The polyQ proteins, complexed with ubiquitin, form inclusions in the neuronal nuclei and cytoplasm, which can be detected by immunohistochemistry. The pathogenesis of neurodegeneration is unknown. It may have to do with dysfunction of the mutated proteins or with diminished proteolytic capacity of the ubiquitin-proteasome system. It has been proposed that polyglutamine repeats bind to and interfere with thefunction of inositol triphosphate receptor type 1 (ITPR1), which is the main excitatory neurotransmitter receptor in Purkinje cells.
Cerebellar ataxias. The pathology in cerebellar cortical (cerebello-olivary) degeneration consists of loss of Purkinje cells and inferior olivary neurons. Loss of Purkinje cells for whatever reason causes transsynaptic degeneration of the inferior olives. In Olivopontocerebellar atrophy (OPCA) there is, in addition, loss of neurons in the pontine nuclei, and atrophy of the transverse fibers of the pons and middle cerebellar peduncles.
OPCA can be viewed as an anatomical sequence which is a component of several diseases. The cerebellum receives sensory input from the nuclei pontis, which convey signals from the cerebral cortex via the middle cerebellar peduncles, and from the inferior olives. When the cerebellar cortex degenerates, both these structures degenerate with it, hence OPCA.
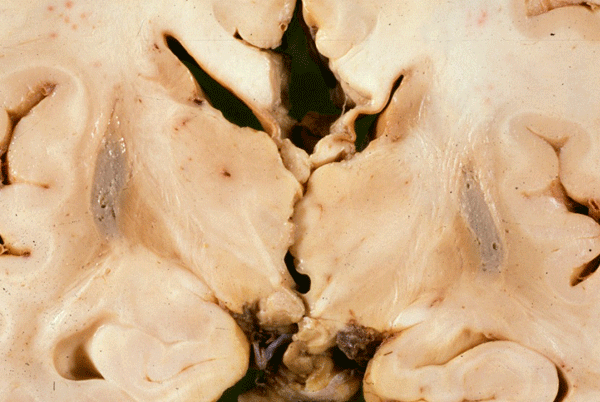 MSA-OPCA |
 MSA-OPCA |
The best known disease entity with a component of OPCA is multiple system atrophy-MSA. MSA is an adult onset, sporadic, progressive neurodegenerative disease characterized clinically by a combination of parkinsonism, ataxia, and autonomic dysfunction. Neuropathological examination reveals loss of normal pigmentation in the substantia nigra, OPCA (atrophy of the cerebellum, basis pontis, and inferior olives), and atrophy of the putamen, which may have a steele-gray color. Microscopic examination of these structures reveals neuronal loss, axon and myelin degeneration, and gliosis. Similar changes are seen in autonomic nuclei of the hypothalamus, brainstem and spinal cord and in many other nuclei. There are no Lewy bodies in the substantia nigra. A key feature of the pathology is the presence of filamentous α-synuclein-immunoreactive cytoplasmic inclusions in oligodendrocytes (glial cytoplasmic inclusions-GCI). GCIs have variable shapes and are located adjacent to oligodendrocyte nuclei. The density of GCIs does not correlate with neuronal loss in various systems. The pathogenesis of MSA assumes a central role for α-synuclein but how this presynaptic neuronal protein is expressed in oligodendrocytes and causes neurodegeneration is unclear.
In children, OPCA is a component of several neurometabolic disorders. These include pontocerebellar hypoplasia, a group of severe autosomal recessive disorders of prenatal onset that cause also degeneration of motor neurons and other systems, congenital glycosylation disorders (previously called carbohydrate deficient glycoprotein syndromes), and other entities.
Other ataxias. Ataxia-telangiectasia (A-T) is a childhood disease characterized by ataxia, extrapyramidal dysfunction, peripheral neuropathy and other neurologic deficits, vascular dilatation, and immunodeficiency. It is caused by mutations of a gene that regulates the cell cycle. These mutations result in defective DNA repair. In addition to cerebellar deegeneration, there is loss of anteriorimag horns, degeneration of brainstem nuclei, substantia nigra, and other neuronal groups, and loss of dorsal root ganglionic neurons with dorsal column degeneration. A-T patients frequently develop opportunistic infections and B-cell lymphomas.
Further Reading
- Pandolfo M. Fiedreich ataxia. Arch Neurol 2008;65:1296-1303. PubMed
- Schorge S, van de Leemput J, Singleton A, et al. Human ataxias:a genetic dissection of inositol triphosphate receptor (ITPR1)-dependent signaling. Trends in Neurosciences 2010;33:211-19. PubMed.
- Namavar Y, Barth PG, Poll-The BT, Baas F. Classification, diagnosis and potential mechanisms in pontocerebellar hypoplasia. Orphanet J Rare Dis. 2011 Jul 12;6:5 0. doi: 10.1186/1750-1172-6-50. PubMed
- Ahmed Z, Asi YT, Sailer A, et al. The neuropathology, pathophysiology and genetics of multiple system atrophy. Neuropathol Appl Neurobiol. 2012;38:4-24. PubMed
- Koeppen AH, Mazurkievicz JE. Friedreich Ataxia: Neuropathology Revised. J Neuropathol Exp Neurol 2013;72(2):78-90. PubMed
- Cook A, Giunti P. Friedreich’s ataxia: clinical features, pathogenesis and management. Br Med Bull 2017124(1): 19–30. PubMed
Updated: June, 2020