MISCELLANEOUS METABOLIC DISORDERS
MENKES DISEASE
Menkes disease is an X-linked metabolic disorder caused by mutations of the ATP7A gene, located on Xq21.1. This gene is expressed in the placenta, intestine, and brain. Its product is important for transport of copper across the placenta, absorption of copper in the small intestine, getting copper across the blood-brain barrier, and moving intracellular copper from the cytosol to several copper-dependent enzymes in the Golgi apparatus. ATP7A is also involved in eliminating excess copper from cells. Thus, mutations of ATP7A cause generalized copper deficiency resulting in severe progressive pathological changes. The clinical manifestations of Menkes disease are due to deficiency of copper-dependent enzymes that are involved in cellular respiration, antioxidant function, neurotransmitter synthesis, the structure and pigmentation of hair, collagen and elastin formation, and other processes.
 Menkes disease |
Menkes disease is characterized by neurological and connective tissue abnormalities. Patients are normal in the first 2 or 3 months of life but then they develop failure to thrive, hypothermia, hypotonia, seizures, severe psychomotor retardation, and usually die before 2 years of age. Their hair is sparse, coarse, steely, and depigmented, and appears twisted (hence Menkes’ kinky hair disease) or broken. Their skin is lax, and their cheeks are redundant. X-rays reveal wormian cranial bones, osteoporosis, and metaphyseal dysplasia. The MRI shows defective myelination, cortical atrophy, ventricular enlargement, and vascular tortuosity. Subdural hematomas often develop as a result of brain atrophy. Serum copper and ceruloplasmin levels are low, and plasma and CSF catecholamines are also low (dopamine-beta-hydroxylase, which is important for catecholamine synthesis, is a copper-dependent enzyme). Milder mutations of ATP7A cause the occipital horn syndrome (OHS; previously known as X-linked cutis laxa), which is characterized mainly by connective tissue abnormalities (hyperelastic skin, loose joints, skeletal abnormalities) and minor neurological abnormalities, or X-linked spinal muscular atrophy. The phenotype of OHS can be explained by deficiency of lysyl oxidase. Copper homeostasis abnormalities are also found in Parkinson’s disease.
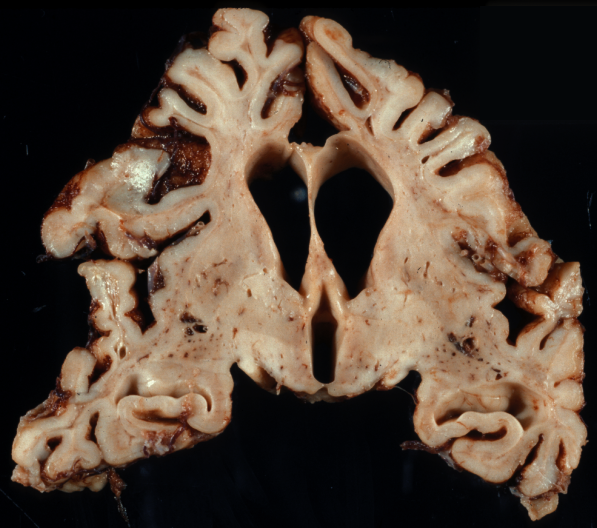 MD. Brain atrophy Menkes disease. Brain atrophy. |
 MD. Cortical degeneration Menkes disease. Laminar degeneration of the cerebral cortex. |
 MD. Subdurals Menkes disease. Organized subdurals enveloping the whole brain. |
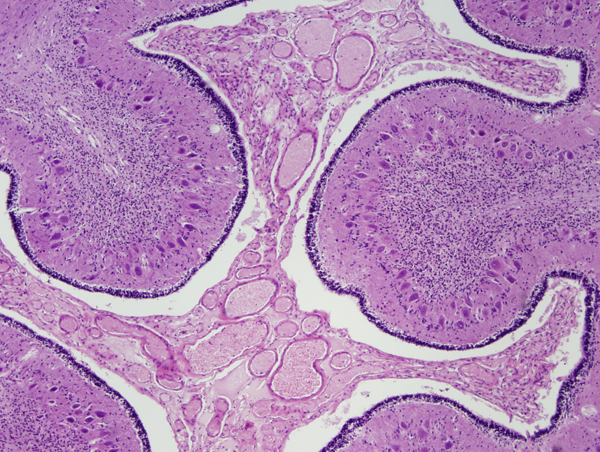 MD. Cerebellar degeneration Menkes disease, cerebellum. Granular neurons are lost but Purkinje cells are relatively spared. Note also the vascular proliferation in the subarachnoid space, a characteristic feature of MD. |
 MD. Cerebellar degeneration |
The brain in Menkes disease shows cerebral and cerebellar atrophy, bilateral chronic subdural hematomas, and tortuous, thin-walled arteries. The cortex shows neuronal loss, mineralized neurons, and gliosis. The white matter is reduced in volume, attenuated, and gliotic. Neuronal loss and gliosis are seen in the thalamus and other subcortical nuclei. The cerebellum shows severe depletion of granular neurons with relative preservation of Purkinje cells, a pattern opposite to that seen in other neurodegenerations. Purkinje cell dendrites branch in the molecular layer forming cactus-shaped expansions. The highest concentration of ATP7A is in the cerebellum and choroid plexus. The neuropathological changes can be attributed, in large part, to deficiency of cytochrome c oxidase, and in that sense have some similarity to mitochondrial disorders.
WILSON'S DISEASE
Wilson’s disease (WD), a.k.a. hepatolenticular degeneration, is an autosomal recessive disease characterized by accumulation of copper in the liver, brain, and other tissues. Copper is a cofactor of several important enzymes. The liver is the key organ for copper homeostasis. WD is caused by mutations of ATP7B on 13q14. The product of this gene is an ATP transporter which is important for biliary copper excretion and incorporation of copper into ceruloplasmin which is the form of copper that circulates in the blood. In the course of WD, the capacity of hepatocytes to process copper is overcome, leading to accumulation of toxic, non-ceruloplasmin-bound copper in hepatocytes and in the circulation. Excess copper accumulating in the liver causes hepatocellular injury that can progress to cirrhosis. More toxic copper is leaked from injured hepatocytes. Toxic copper is taken up by the brain, especially the basal ganglia (lenticular nucleus; hence hepatolenticular degeneration), and other organs and tissues. The neurological manifestations of WD consist of movement disorders, dystonia, and psychiatric symptoms. These are due to damage of the basal ganglia and other structures. A visible marker of WD is the Kayser-Fleischer ring, a brownish band caused by copper deposition at the outer edge of the cornea. The neuropathological changes range from rarefaction to frank necrosis with prominent Alzheimer type 2 astrocytes and involve the basal ganglia, thalamus, cerebellum and brainstem. Repeated bouts of hepatic encephalopathy can also cause similar symptoms and pathology.
Further Reading
- Lee CE, Singleton KS, Wallin M, Faundez V. Rare Genetic Diseases: Nature's Experiments on Human Development. iScience 2020 May 22;23(5):101123. doi: 10.1016/j.isci.2020.101123. PubMed
Updated: September, 2023