MITOCHONDRIAL DISORDERS
ENERGY PRODUCTION AND FREE RADICALS
This page describes conditions that impair oxidative phosphorylation (oxphos), the process by which mitochondria capture the energy in pyruvate (produced by carbohydrate glycolysis) and fatty acids and store it as ATP. The end product of pyruvate dehydrogenation and fatty acid beta oxidation, acetyl-CoA, is oxidized in the Krebs cycle. Electrons generated in the Krebs cycle and during beta oxidation flow through the respiratory chain (electron transport chain-ETC). The respiratory chain consists of five complexes of transmembrane proteins, located on the inner mitochondrial membrane. Complexes I-IV pump protons out of the mitochondrial matrix, building a proton gradient. Then protons reverse flow and pass through complex V generating ATP. A small proportion of oxygen that enters the respiratory chain is converted to toxic byproducts, oxygen radicals (free radicals). Most of these are detoxified by protective cellular enzymes and vitamin antioxidants. If they are not neutralized, free radicals can damage lipids, proteins, and nucleic acids. This damage is more severe at the site of free radical generation, i.e., the mitochondria. Mitochondria are deficient in protective cellular mechanisms, most of which reside in the cytosol. Respiratory chain dysfunction increases free radical production. This creates a vicious cycle, which increases mitochondrial damage and causes worse respiratory chain dysfunction and more free radical generation. Cellular damage in mitochondrial disorders is due to free radicals and energy deficiency. Both these factors can also trigger necrosis and apoptosis.
GENETICS OF MITOCHONDRIAL DISORDERS
The
respiratory chain consists of 90 proteins. Seventy seven of these are encoded by nuclear DNA (nDNA), synthesized in the RER, and imported into the mitochondria. The other 13 are encoded by mitochondrial DNA (mtDNA) and are synthesized in the mitochondria. Each mitochondrion contains two to ten copies of the mitochondrial genome, a 16.6 kb double-stranded circular DNA molecule, attached to the inner mitochondrial membrane. In addition to the 13 respiratory chain protein genes, the mitochondrial genome contains 2 genes encoding rRNAs and 22 genes encoding tRNAs, 37 genes in all.
Respiratory chain disease can be caused by either mtDNA or nDNA mutations. The mtDNA mutations are (1) large scale deletions; (2) point mutations of rRNA and tRNA genes; and (3) point mutations of protein-coding genes. Large scale deletions of mtDNA and point mutations of tRNA and rRNA genes affect protein synthesis and have a profound effect on all respiratory chain complexes (except II, all 4 subunits of which are encoded by nDNA.)
TYPE-EFFECT
OF MUTATION |
GENES |
SYNDROME |
PHENOTYPE |
RRFs |
|---|---|---|---|---|
| Deletions | CPEO, KSS,PS | Present | ||
| tRNA, rRNA point mutation | tRNA leucine tRNA lysine |
MELAS MERRF |
Present | |
| Protein-coding mutation | Complex I subunits ATP synthase subunit 6 |
LHON NARP |
Absent |
KSS: Kearns-Sayre Syndrome
CPEO: Chronic Progressive External Ophthalmoplegia
LHON: Leber Hereditary Optic Neuropathy
MELAS: Mitochondrial Encephalomyopathy with Lactic
Acidosis and Strokelike Episodes
MERRF: Myoclonic Epilepsy with Ragged Red Fibers
NARP: Neuropathy, Ataxia, And Retinitis Pigmentosa
PS: Pearson Syndrome
The mtDNA is not autonomous; it depends on nuclear-encoded factors for its replication, transcription, and translation. In addition to encoding most subunits of the respiratory chain, nDNA encodes proteins that are important for respiratory chain assembly, mtDNA maintenance, mitochondrial membrane integrity, and other aspects of mitochondrial function. The nDNA mutations that affect the respiratory chain can be divided into (1) mitochondrial depletion syndromes (MDS); (2) mutations causing multiple mtDNA deletions; and (3) mutations causing isolated ETC deficiencies.
TYPE-EFFECT
OF MUTATION |
GENES |
SYNDROME |
PHENOTYPE |
RRFs |
|---|---|---|---|---|
| MDS | POLG, DGUOK TK2, SUCLA2 |
Alpers, Myopathy |
Present in myopathy | |
| mtDNA deletion | ANT1, PEO1, POLG TP |
CPEO, ARCO, MNGIE |
Absent in MNGIE Present in myopathy |
|
| ETC subunit-complex deficiency | COX assembly genes | LS, myopathy | Absent in LS Present in myopathy |
ARCO: Autosomal Recessive
Cardiomyopathy, Ophthalmoplegia
CPEO: Chronic Progressive External Ophthalmoplegia
LS: Leigh Syndrome
MNGIE: Mitochondrial Neurogastrointestinal Encephalomyopathy
NARP: Neuropathy, Ataxia, And Retinitis Pigmentosa
Nuclear gene defects are transmitted in a mendelian fashion: most are autosomal recessive. Because the ovum has numerous mitochondria and the sperm almost none, mutations of mitochondrial genes are transmitted through the mother (maternal inheritance) or are sporadic. Nuclear gene defects affect all cells equally. Defects of mtDNA affect cells unevenly. Each cell contains 2 copies of any given nuclear gene but thousands of (mitochondria and) copies of mitochondrial genes. Because of the random way in which mitochondria segregate in dividing cells, wild type and mutant mtDNA coexist in variable proportions in any given cell, a phenomenon called heteroplasmy. In nondividing cells, such as myocytes and neurons, this proportion is relatively stable. In dividing cells, it may shift such that, after several cell cycles, a given cell may come to contain mostly mutant mtDNA (mitotic segregation). Cellular dysfunction develops when the proportion of mutant mtDNA exceeds a certain threshold, typically 80%-90%. Thus, in mtDNA mutations, the genetic defect is dynamic, and cell and tissue dysfunction is in a state of flux. Consequently, the severity of disease in any given cell line cannot be predicted and the clinical phenotype shows great variability. With some exceptions, most mtDNA mutations are heteroplasmic, presumably because homoplasmy for mutant mtDNA would be lethal.
CLINICAL ASPECTS OF MITOCHONDRIAL DISORDERS
Over 270 genetic entities of mitochondrial disorders
have been recorded. They affect virtually all organ
systems and cause hepatic, gastrointestinal, renal,
hematopoietic, and endocrine abnormalities. However,
the cells and organs that are most severely affected
are those that have the highest energy consumption,
namely the brain and skeletal and cardiac muscle (mitochondrial
encephalomyopathies). The major mitochondrial
disorders have distinct core phenotypes but show also
markedly varied and overlapping clinical features.
Some mitochondrial disorders, e.g., Leber Hereditary
Optic Neuropathy, affect a single organ. Most
cause multi-organ dysfunction with prominent neurological
abnormalities and muscle disease.
The neurological abnormalities include
loss of vision and hearing, migraine headaches, seizures
and myoclonus, focal neurological deficits, encephalopathy,
psychomotor retardation, dementia, ataxia, spasticity,
motor neuron disease, system degenerations, and peripheral
neuropathy. Muscle disease may present
with weakness, exercise intolerance, rhabdomyolysis,
myopathic face, chronic fatigue, a fibromyalgia-like
picture, and an abnormal EMG. Extraocular muscles are especially
susceptible because they have a high proportion of
type 1 (oxidative) fibers. Thus, ptosis and ophthalmoplegia
are very common in mitochondriopathies.
The neuroimaging findings of mitochondrial disorders include abnormal signal in the basal ganglia, basal ganglia calcification, cerebral and cerebellar atrophy, bilateral striatal necrosis, cerebellar hypoplasia, infarcts, and leukoencephalopathy.
LABORATORY DIAGNOSIS OF MITOCHONDRIAL DISORDERS
The laboratory investigation of mitochondrial disorders includes determination of lactic acid and lactate/pyruvate ratio in blood and CSF, a muscle or other tissue biopsy, enzyme analysis of muscle tissue for respiratory chain defects, and DNA analysis. Many children with unexplained encephalopathy and acidosis are suspected to have mitochondrial or organic acid disorders and are subjected to extensive laboratory tests, including muscle biopsies. Most of the time, this work-up is negative. It is worth pointing out that the clinical manifestations and MRI findings of metabolic disease are nonspecific and the most common causes of lactic acidosis are hypoxic-encephalopathy and sepsis.
Biochemical findings
The key laboratory test for diagnosis of mitochondrial disorders is determination of blood and CSF lactate. In mitochondrial disorders, unprocessed pyruvate is converted to lactate. Both lactate and pyruvate are elevated, and the L/P ratio increases. Elevation of blood and CSF lactate above 2.5 mM/dL and elevation of the L/P ratio (if lactate is high) indicate mitochondrial disease. Alanine, glycine, and other amino acids may also be elevated.
 Hepatic steatosis in mitochondrial disease |
 Mitochondria in hepatocyte |
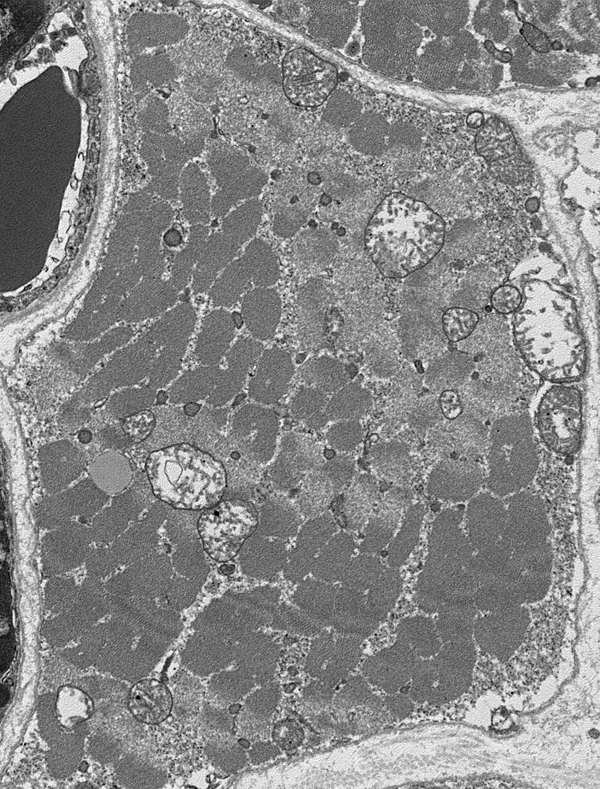 Large mitochondria in myocyte |
Most patients with mitochondrial disorders have muscle biopsies, which are typically processed for enzyme histochemistry (including NADH-tetrazolium reductase, Succinic Dehydrogenase, Cytochrome C Oxidase, Gomori trichrome) and electron microscopy (see below). Mitochondrial accumulation and hypertrophy are compensatory abnormalities and increase with age, so changes may be mild and RRFs may be absent in children. Hepatic dysfunction from mtd is seen primarily in children. Liver biopsy may show steatosis, abnormal mitochondria, and nonspecific abnormalities.
Enzyme analysis of the ETC.
Muscle, heart, or liver tissue frozen in liquid nitrogen can be used for spectrophotometric analysis of the ETC. Polarographic analysis of ETC enzymes can also be done on mitochondria-enriched fractions derived from fresh (not frozen) muscle tissue. Enzyme analysis can also be done on lymphocytes isolated from blood within 24 hours of collection.
DNA analysis
Whole exome (or other) sequencing of DNA extracted from blood can be used to detect mutations of nuclear genes that affect mitochondrial function. Next-generation sequencing of the mitochondrial genome in blood DNA can be used to detect mtDNA mutations. DNA analysis of tissues such as muscle and liver can be used for diagnosis of mtDNA gene mutations. Tissue testing picks up tissue-specific mutations and excludes situations of low-level heteroplasmy in blood in which mtDNA mutations may be undetectable. Renal epithelial cells have a high content of mtDNA, and DNA analysis of urine can be used for diagnosis of MELAS.
PATHOLOGY OF MITOCHONDRIAL DISORDERS
Several mitochondrial disorders are accompanied by a massive proliferation and enlargement of mitochondria in myofibers. On electron microscopic examination, these mitochondria are large and structurally abnormal. They have a concentric or other unusual arrangement of their cristae and some of them contain crystal-like inclusions.
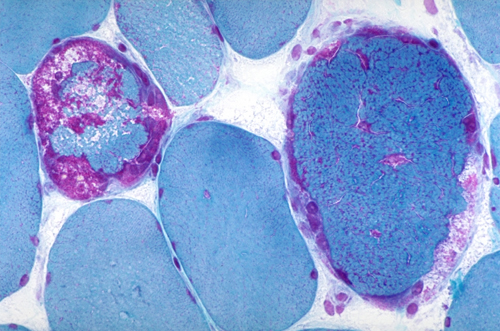 Ragged red fibers |
 Subsarcolemmal mitochondrial aggregates |
 COX-negative fibers |
 Abnormal mitochondria in RRF |
These mitochondrial clusters appear as red deposits in cryostat sections of muscle stained with modified Gomori trichrome. Myofibers with such deposits are called ragged red fibers (RRFs)-ragged because their structure is disrupted by the accumulating mitochondria. RRFs are the hallmark of mitochondrial disorders and occur in no other metabolic disease but are only present in about one-third of them. They may be few or may be the majority of myofibers in a biopsy. Thus absence of RRFs does not rule out a mitochondrial disorder. RRFs occur in large deletions and tRNA mutations and in mutations of nDNA that cause multiple mtDNA deletions or reduction of mtDNA copy number. The common denominator of these conditions is impairment of intramitochondrial protein synthesis. They are less abundant or absent in mutations involving protein-coding genes. RRFs are Succinic Dehydrogenase (SDH) and NADH hyperreactive. The SDH reaction product is deep blue, hence RRFs are also ragged blue fibers. SDH (complex II) is entirely encoded by nDNA and a good marker of mitochondrial proliferation. Cytochrome C Oxidase (COX) is a component of complex IV which is encoded in part by mtDNA and in part by nDNA. COX is synthesized in mitochondria and reflects respiratory chain function. Therefore, RRFs in mutations affecting protein synthesis are deficient in COX activity. RRFs occurring in mutations of individual respiratory chain genes (except for those that encode COX components) are generally COX-positive. RRFs are rare in children with mitochondrial disorders and occur in other muscle diseases-notably inclusion body myositis and polymyalgia rheumatica. In these conditions, they are thought to be due to mtDNA damage induced by free radicals. A few RRFs are also seen incidentally in the muscles of older people. Milder mitochondrial proliferation often takes the form of subsarcolemmal aggregates that do not disrupt myofiber structure.
The CNS pathology of mitochondrial disorders (see below) affects gray and white matter. Gray matter lesions consist of hypoxic-ischemic neuronal changes affecting individual or groups of neurons (MELAS), neuronal loss (MERRF), and vacuolization and vascular proliferation of the neuropil with relative sparing of neurons (LS). The white matter pathology is vacuolating myelinopathy, seen mainly in the KSS.
LEIGH SYNDROME (LS)
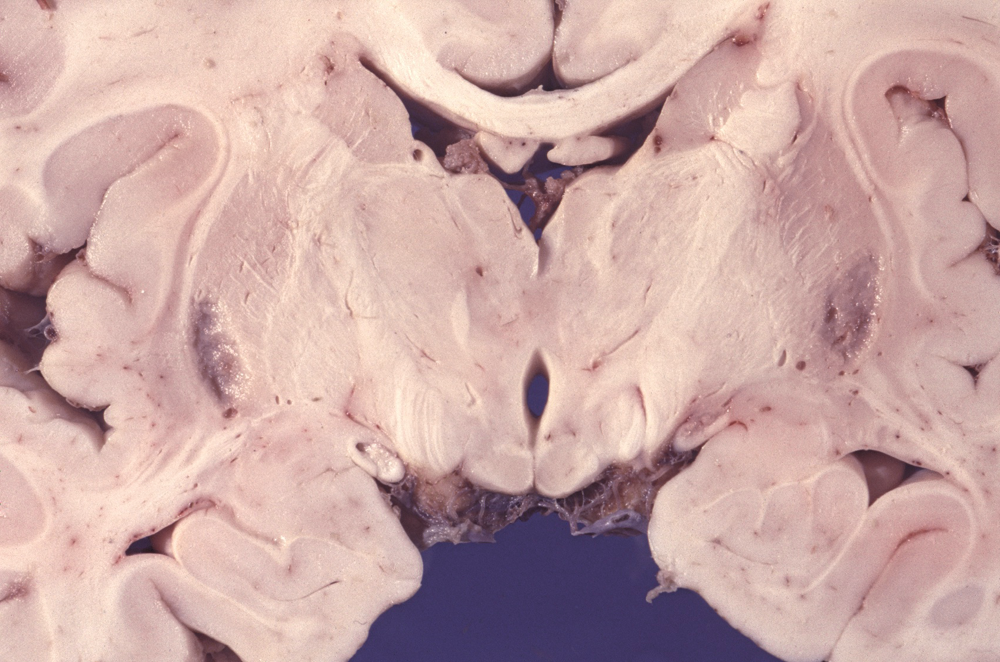 Leigh syndrome. Basal ganglia. |
 Leigh syndrome. Brainstem. |
 Leigh syndrome. Medulla. |
Definition and clinical findings: A progressive childhood mitochondrial encephalopathy that damages primarily the brainstem and basal ganglia causing developmental delay, hypotonia, ophthalmoplegia, nystagmus, ataxia, and other symptoms. The heart, liver, kidneys, and other organ systems may also fail. After a period of normal development, symptoms usually appear by 2 years, frequently coinciding with an infection. Mean age at death is 5 years. LS is the most common mitochondrial disorder in children.
Genetics: LS is genetically diverse. It is caused by mutations of more than 75 mitochondrial and nuclear genes that encode OXPHOS subunits, pyruvate dehydrogenase components, and other enzymes involved in energy metabolism. The net effect of these mutations is severe ATP depletion. Most cases are autosomal recessive, but X-linked and maternal inheritance are seen in some cases.
Pathological and imaging findings: Congestion, softening, and atrophy of basal ganglia and brainstem tectum and tegmentum. The cerebellar dentate nucleus, thalamus, and spinal cord may also be affected. Attenuation of the neuropil, spongiosis, and vascular proliferation with relative preservation of neurons. The topography of the lesions is somewhat similar to the Wernicke-Korsakoff syndrome. The MRI shows hyperintense T2 lesions with increased lactate signal. No ragged red fibers are seen in most cases.
KEARNS-SAYRE SYNDROME (KSS) AND CHRONIC PROGRESSIVE EXTERNAL OPHTHALMOPLEGIA (CPEO).
Definition and clinical findings: A mitochondrial disorder caused by large mtDNA deletions, characterized clinically by ophthalmoplegia, weakness, ataxia, pigmentary retinopathy, loss of hearing, dementia, and seizures. The numerous non-neurological manifestations of KSS include hypertrophic and dilated cardiomyopathy, cardiac conduction abnormalities, impaired GI motility, diabetes mellitus and other endocrine abnormalities, short stature, and renal dysfunction.
Genetics: Most KSS and CPEO cases are sporadic and are caused by new mtDNA deletions that wipe out a large amount (up to 50%) of the mitochondrial genome including tRNA genes, thus affecting mitochondrial protein synthesis.
Pathological findings: RRFs and spongy myelinopathy
MITOCHONDRIAL ENCEPHALOMYOPATHY WITH LACTIC ACIDOSIS AND STROKELIKE EPISODES (MELAS)
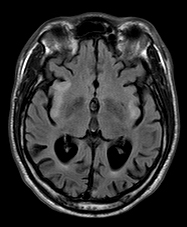 MELAS |
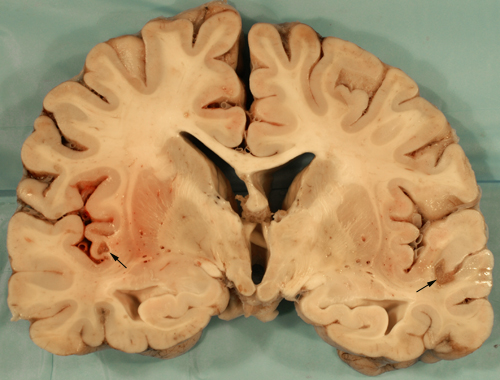 MELAS. Small infarct. |
Definition and clinical findings: A mitochondrial disorder characterized by encephalopathy (seizures, dementia), recurrent stroke-like episodes at a young age, myopathy, and lactic acidosis. Ataxia, deafness, pigmentary retinopathy, and short stature are seen in some patients.
Genetics: Maternally inherited mtDNA mutations involving the tRNA leucine gene cause 80% of MELAS cases.
Pathological findings: COX-positive RRFs, cortical and subcortical infarct-like lesions, pseudolaminar necrosis, and basal ganglia mineralization
MYOCLONIC EPILEPSY WITH RAGGED RED FIBERS (MERRF).
Definition and clinical findings: A mitochondrial disorder characterized by myoclonus, epilepsy, ataxia, and dementia.
Genetics: Maternally inherited mtDNA mutations affecting the tRNA lysine gene cause 90% of MERRF cases.
Pathological findings: RRFs and system degenerations involving cerebellar, brainstem, and spinal cord tracts and nuclei
ALPERS-HUTTENLOCHER SYNDROME AND POLG RELATED DISORDERS
Mitochondrial DNA is replicated and repaired by DNA polymerase γ, the catalytic subunit of which is encoded by POLG, a nuclear gene on 15q25. POLG mutations (the most common is the A467T mutation) cause several syndromes that affect the CNS, vision and hearing, peripheral nerves, skeletal and cardiac muscle, liver, gastrointestinal tract, and other organs. The major clinical syndromes associated with POLG mutations are autosomal recessive and autosomal dominant progressive external ophthalmoplegia (PEO), childhood myocerebrohepatopathy spectrum (MCHS), myoclonus epilepsy myopathy sensory ataxia (MEMSA), ataxia-neuropathy spectrum (ANS), and Alpers-Huttenlocher syndrome (AHS). These syndromes overlap with and recapitulate the findings of mitochondrial disorders due to mtDNA and other nDNA abnormalities. AHS is a fatal syndrome presenting in infancy and characterized by encephalopathy, seizures, headaches, and hepatic failure, which occurs spontaneously or is triggered by valproic acid. Hepatic failure following valproic acid administration may also develop in individuals without AHS who carry POLG polymorphisms. The brain in AHS shows nonspecific changes (neuronal degeneration, neuronal loss, and gliosis), which affect initially the cerebral cortex and later deep nuclei and the brainstem. The liver shows nonspecific findings including macro- and microvesicular steatosis, bile duct proliferation, and hepatocyte loss leading to fibrosis and cirrhosis.
LEBER HEREDITARY OPTIC NEUROPATHY (LHON) AND AUTOSOMAL DOMINANT OPTIC ATROPHY (DOA)
 LHON |
Definition and clinical findings: A mitochondrial disorder that causes painless progressive loss of central vision. Some LHON patients also have dystonia, pseudobulbar palsy, intellectual deterioration, and muscle weakness. Many patients have the Wolff-Parkinson-White syndrome. Such cases are referred to as “LHON plus” phenotypes. Female LHON patients may have multiple sclerosis-like manifestations. DOA is more common than LHON. It begins insidiously in childhood, sometimes at birth and causes central visual loss and impaired color vision. It has a highly variable expression and course but severe visual loss develops in most patients eventually. Fundoscopic examination reveals temporal pallor of the optic disc. Similar to LHON, some DOA patients have an extended phenotype-“DOA plus”-with additional neuromuscular manifestations including a multiple sclerosis-like illness.
Inheritance: LHON is maternally inherited and shows a male prevalence. It is caused by mtDNA mutations that encode subunits of ETC complex I. DOA is autosomal dominant.
Pathological findings: Loss of retinal ganglion cells in the perifoveal region (macula densa) and degeneration of the papillomacular bundle. There are no RRFs.
Mitochondrial disorders are progressive and show significant clinical and genetic variability and overlap. Some phenotypes are caused by defects of mtDNA, others by defects of nDNA, and some by both. Similar syndromes can be caused by a variety of mutations and any given mutation may have a variable phenotype.
The spectrum of genetic mitochondrial disease in neurology and medicine is expanding. Additionally, mitochondrial DNA damage from free radicals can develop in absence of a genetic disorder. Because mitochondria replicate even in postmitotic cells such as neurons, such damage is propagated intracellularly and may get worse with time. Mitochondrial DNA damage and free radicals have been implicated in the pathogenesis of HIE, neurodegenerative diseases, and the gradual deterioration of tissues that occurs with advancing age.
Further Reading
- DiMauro S, Davidzon G. Mitochondrial DNA and disease. Ann Med 2005; 37:222-32. PubMed
- Morava E, van den Heuvel l, Hol f et al. Mitochondrial disease criteria. Diagnostic applications in children. Neurology 2006;67:1823-6. PubMed
- DiMauro S, Schon EA. Mitochondrial Disorders in the Nervous System. Annu Rev Neurosci 2008;31:91-123. PubMed
- Haas RH et al. The in-depth evaluation of suspected mitochondrial disease. TheMitochondrial Medicine Society’s Committee on Diagnosis. Molec Genet Metab 2008;94:16-37. PubMed
- DiMauro S. Mitochondrial encephalomyopathies-Fifty years on. Neurology 2013;81:281-91. PubMed
- Lake NJ, Bird MJ, Isohanni P, Paetau A. Leigh Syndrome: Neuropathology and Pathogenesis. J Neuropathol Exp Neurol 2015;74:482-492. PubMed
- Leigh Syndrome: One disorder, more than 75 monogenic causes.Lake NJ, Compton AG, Rahman S, Thorburn DR. Ann Neurol. 2015 Oct 27. doi: 10.1002/ana.24551. [Epub ahead of print] Review.PMID: 26506407
Updated: May, 2017