LEUKODYSTROPHIES
Leukodystrophies-LDs (from the Greek words leukos: white and dystrophia: faulty development, degeneration) are genetic metabolic disorders of the white matter that affect glial cells. In LDs, myelin may be absent or decreased, of an abnormal structure, and unstable. Normal myelination may not be attained and what myelin is formed may break down. Loss of myelin leads to loss of axons. Some LDs affect myelin and myelin producing cells. However, all cells that reside in the white matter are important for myelin structure and maintenance. Consequently, a LD may arise from genetic abnormalities involving other than myelin-producing cells. Myelin is composed of about 70% lipid and 30% protein. CNS and peripheral nervous system (PNS) myelin have similar lipids but significantly different proteins. For this reason, LDs involving myelin lipids tend to affect the CNS and the peripheral nervous system (PNS) while LDs caused by myelin protein abnormalities affect the CNS only. Some LDs affect other cells, e.g., astrocytes, and disrupt other biochemical pathways resulting in indirect damage to myelin. LDs usually present in children and young individuals with spasticity, ataxia, cognitive decline and seizures, but they have a wide clinical spectrum ranging from fatal congenital disease to milder adult presentations. The best known LDs are listed in the table below. Some of these are described in the chapters of lysosomal and peroxisomal disorders. A brief account of some other LDs is given in this page.The term Leukoencephalopathy (LE) refers to white matter abnormalities in general. Acquired LE occurs in chronic ischemia of the white matter and is associated with leukoaraiosis (rarefaction), i.e., hypodensity of the white matter on CT and increased signal on T2 or FLAIR MRI. Binswanger encephalopathy, a white matter degeneration that has been linked to hypertension, is a pathological counterpart of some cases of leukoaraiosis. Some LEs have a genetic basis and are secondary to neuronal, vascular or other pathology. Genetic LEs may be associated with lysosomal storage, mitochondrial, amino acid and other disorders. This group includes also the following genetic vasculopathies: Cerebral Autosomal Dominant Arteriopathy with Subcortical Infarcts and Leukoencephalopathy (CADASIL), Cerebral Autosomal Recessive Arteriopathy with Subcortical Infarcts and Leukoencephalopathy (CARASIL), COL4A1-related small-vessel disease and Cerebral Amyloid Angiopathy. In these conditions, chronic white matter ischemia resulting from vascular narrowing causes loss of white matter, resulting in abnormal MRI signal that may mimic LE. White matter infarcts and hemorrhages may also occur.
| LEUKODYSTROPHY | CELLULAR DEFECT | BIOCHEMISTRY | GENETICS |
|---|---|---|---|
| MLD | Oligodendroglia/SC Lysosomal storage |
ASA deficiency Sulfatide storage |
Autosomal recessive |
| Globoid cell leukodystrophy | Oligodendroglia/SC Lysosomal storage |
GALC deficiency Galactocerebroside storage | Autosomal recessive |
| XALD/AMN | Oligodendroglia/SC Peroxisomal | ALDP deficiency VLCFA accumulation | X-linked |
| PMD/SPG2 | Oligodendroglia protein synthesis | PLP1 abnormality | X-linked |
| Alexander disease | Astrocyte protein synthesis |
GFAP overproduction | Autosomal recessive |
| Canavan disease | Neurons, oligodendroglial precursors | Amino acid metabolism ASPA deficiency, NAA accumulation |
Autosomal dominant |
| KSS | Mitochondrial | Unknown | Maternal inheritance |
| Amino-acid disorders | Amino acid metabolism | Unknown | Most Autosomal recessive |
ALDP: ALD protein; AMN: adrenomyeloneuropahty; ASA: arylsulfatase A; ASPA:Aspartoacylase; GALC: galactocerebrosidase; GFAP: glial fibrillary acidic protein; KSS: Kearns-Sayre syndrome; MLD: metachromatic leukodystrophy; NAA: n-acetyl N aspartate; PLP1:proteolipid protein 1; PMD: Pelizaeus-Merzbacher disease; SC: Schwann cell; SPG2: spastic paraplegia type 2; VLCFA: very long chain fatty acids; XALD: X-linked adrenoleukodystrophy
METACHROMATIC LEUKODYSTROPHY (see lysosomal disorders)
GLOBOID CELL LEUKODYSTROPHY-Krabbe disease (see lysosomal disorders)
X-LINKED ADRENOLEUKODYSTROPHY (see peroxisomal disorders)
PELIZAEUS-MERZBACHER DISEASE (PMD).
PMD is a rare X-linked leukodystrophy due to abnormalities of the major CNS myelin protein, proteolipid protein 1 (PLP1) and its spliced isoform DM20. It is caused by duplications, deletions, and point mutations of the PLP1 gene, which is located on Xq22. PLP1-related disorders have a wide clinical spectrum which correlates with the underlying molecular defect.
 Pelizaeus-Merzbacher disease |
 PMD-cerebellar degeneration |
Classic PMD presents in the first year of life with delay in motor development, nystagmus, ataxia, spasticity, and abnormal movements. The white matter is reduced in volume and shows increased signal intensity on FLAIR MRI images. Death occurs from childhood to adult age. A milder variant, spastic paraplegia type 2 (SPG2) presents later with weakness and spasticity of the lower extremities. Female carriers of the disease are usually normal. Some females with point mutations may have mild symptoms. The pathology varies with the severity and stage of the disease. There is loss of myelin and oligodendrocytes sparing the subcortical fibers. Many cases show a characteristic patchy (tigroid) pattern of myelin loss. Peripheral nerves are normally myelinated. A large proportion of patients with a PMD phenotype have no PLP1 abnormalities. Some of these patients have an autosomal recessive Pelizaeus-Merzbacher-like disease (PMLD) which is caused by mutations of the GJC2 gene that disrupts SOX10 activity (see further on.)
LEUKOENCEPHALOPATHY WITH VANISHING WHITE MATTER (VWM), also known as CHILDHOOD ATAXIA WITH CENTRAL HYPOMYELINATION
VWM is a recently recognized autosomal recessive leukoencephalopathy (previously reported as orthochromatic sudanophilic leukodystrophy) which causes ataxia and spasticity and, less frequently, gognitive deterioration, visual impairment and seizures. In its classic form, it affects children and and is slowly progressive with episodes of neurologic deterioration triggered by physical (febrile illness, head trauma) or emotional (fright) stress. It is a fatal disorder.
 Vanishing white matter disease |
 Advanced VWMD |
 Vanishing white matter disease |
Older individuals are also affected and have milder symptoms. The MRI shows progressive water accumulation in the white matter leading to cyst formation. The brain looks normal externally. In sections, the cerebral cortex is normal. The white matter has a gelatinous consistency or is extremely rarefied and cystic. In advance stages, there is severe loss of white matter. Microscopic examination shows loss of myelin and axons, limited numbers of reactive astrocytes with atypical cytological features, and no inflammation. There is a peculiar increase of oligodendrocytes around cavitated areas and some of these oligodendrocytes have vacuolated cytoplasm. In spite of the severe white matter loss, the cortex and gray matter structures are normal.
VWM is caused by mutations of genes encoding any of the 5 subunits of the eukaryotic translation initiation factor 2B (eIF2B), EIF2B1-EIF2B5. eIF2B regulates the initial steps of protein synthesis. This process is particularly critical in response to cellular stress and its malfunction activates cellular pathways that lead to cell cycle arrest and apoptosis. Although eIF2B function is important for all cells and tissues, only the white matter is affected, suggesting a special vulnerability during the phase of central myelin formation. Other aspects of the disease such as the sparing of the cortex and the discrepancy between the severe white matter pathology and the degree of functional impairment are poorly understood.
ALEXANDER DISEASE
Alexander Disease (AD) is a rare leukodystrophy, which is characterized by accumulation of Rosenthal fibers (RF) in astrocytes. Patients with the more common infantile form of AD present in the first 2 years of life with psychomotor retardation, megalencephaly, spasticity and seizures. The less frequent juvenile AD presents with seizures, brainstem dysfunction (dysphagia, dysarthria, hiccups), ataxia, and cognitive deterioration. Adult onset AD is characterized by progressive brainstem dysfunction. MRI shows low T1 and high T2 signal in the white matter, more severe in the frontal lobes.
 Alexander disease |
 Alexander disease. Rosenthal fibers |
The brain, in the typical infantile form of AD is enlarged but appears normal externally. Sections show white matter softening, disintegration, and cavitation. Microscopic examination shows myriads of RFs throughout gray and white matter, more densely concentrated along the pial and ependymal surfaces and around vessels. Along with the RFs, there is loss of myelin and variable, in some cases severe, loss of axons and myelin. Under the electron microscope, RFs appear as granular osmiophilic deposits in astrocytic processes, embedded in intermediate filaments.
{kind=link}
AD is caused by dominant gain-of-function mutations of the GFAP (Glial Fibrillary Acidic Protein) gene, located on17q21. GFAP is the protein of intermediate astrocytic filaments. In AD, GFAP is overproduced and deposited in the astrocytic cytoplasm and processes as RFs. Thus, AD is primarily a disorder of astrocytes. RF numbers correlate with the distribution of fibrillary astrocytes. The pathogenesis of the myelin abnormality is poorly understood. The pathology does not involve the white matter as selectively as other leukodystrophies do. The cortex is extensively involved, explaining the frequency of seizures.
CANAVAN DISEASE (SPONGY LEUKODYSTROPHY)
Canavan disease (CD) is an autosomal recessive disorder that presents usually in the first few months of life with macrocephaly, lack of head control, hypotonia (and later spasticity,) developmental delay and seizures. Neurological development is arrested and patients die in their teens. In early stages, the brain is significantly larger than normal and there is no myelin.
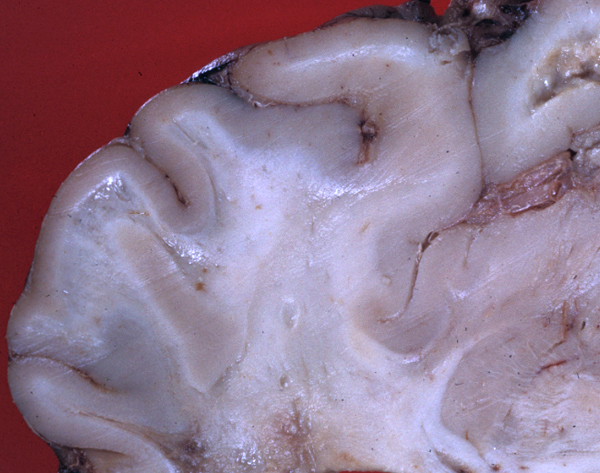 Canavan disease |
 Spongy degeneration |
As the disease advances, brain weight is reduced and white matter atrophy develops. On microscopic examination, numerous vacuoles are seen in the deeper cortical layers, cortex-white matter junction, white matter and cerebellum, imparting a spongy appearance to the affected areas which gave the disease its name. Unlike most other leukodystrophies, the subcortical-U- fibers are affected. CD bears some resemblance to the spongy myelinopathy seen in amino acid and organic acid disorders. These vacuoles appear to develop between myelin lamellae. Oligodendroglial cells and axons are preserved. In advanced CD, myelin is absent and there is no macrophage reaction. No fibrous gliosis is present but there are Alzheimer type 2 astrocytes in the cortex.
CD is caused by mutations of ASPA, which encodes the enzyme Aspartoacylase. Deficiency of aspartoacylase results in build up of its substrate, n-acetyl L-aspartate (NAA) in neurons and oligodendroglial presursors, which leads to neuronal dysfunction and myelin deficiency. ASPA mutations are very common among Ashkenazi Jews but have also been detected in many other populations. The diagnosis of CD can be made by measuring NAA levels in urine, blood and CSF, by assaying aspartoacylase activity in cultured fibroblasts and by molecular methods.
OTHER LEUKOENCEPHALOPATHIES AND RELATED WHITE MATTER LESIONS
AMINO ACID AND ORGANIC ACID DISORDERS
Several amino acid and organic disorders, most notably nonketotic hyperglycinemia, show spongy myelinopathy (SM) as their main or only neuropathologic abnormality. SM is caused by vacuoles developing between myelin lamellae. SM is not associated with myelin break down and is not progressive. The pathogenesis of this lesion is unclear and it is not known if it affects neurological function. Similar changes are seen in CD, mitochondrial disorders, galactosemia, and other conditions.
MITOCHONDRIAL DISORDERS (MTDs)
Some MTDs, especially the Kearns-Sayre syndrome (KSS), affect the white matter extensively. Usually, there is also involvement of the basal ganglia, cortex, and other structures. There are rare MTDs that cause a diffuse white matter abnormality as their main or only CNS manifestation. The pathology in the KSS is spongy myelinopathy due to splitting of myelin lamellae.
SOX10 MUTATIONS
The transcription factor SOX10 is expressed in neural crest cells and oligodendrocytes and is essential for central and peripheral myelin formation and maintenance. SOX10 mutations cause PCWH (peripheral demyelinating neuropathy, central dysmyelinating leukodystrophy, Waardenburg syndrome and Hirshprung disease).
Further Reading
- Boespflug-Tanguy O, Labauge P, Fogli A, et al. Genes Involved in the Leukodystrophies: A Glance at Glial Functions. Curr Neurol Neurosci Rep 2008;8:217-29. PubMed
- Inoue K. PLP 1-related inherited dysmyelinating disorders: Pelizaeus-Merzbacher disease and spastic paraplegia type 2. Neurogenetics 2005;6:1-16. PubMed
- Bugiani M, Boor I, Powers J, et al Leukoencephalopathy with Vanishing White Matter: A Review. J Neuropathol Exp Neurol 2010;69:987-96. PubMed
- Sawaishi Y. Review of Alexander disease: Beyond the classical concept of leukodystrophy. Brain Dev. 2009;31:493-8. PubMed
- Kumar S, Mattan NS, deVellis J. Canavan disease: a white matter disorder. Ment Retard Devel Disabil Res Rev 2006;12:157-65. PubMed
- Lupski J. Interruption of SOX10 function in myelinopathies. Ann Neurol. 2010;68:121-3. PubMed
- van der Knaap, M. S. & Bugiani, M. Leukodystrophies: a proposed classification system based on pathological changes and pathogenetic mechanisms. Acta Neuropathol 2017; 134:351–382 PubMed .
Updated: March, 2019