LSDs CONTINUED-SELECTED DISORDERS
GANGLIOSIDOSES
Sphingolipids consist of a backbone of ceramide (N-acylsphingosine) with various attached side chains. Gangliosides are glycosphingolipids containing sialic acid. They are major constituents of cell membranes, and are especially rich in neuronal membranes. Deficiency of enzymes of sphingolipid degradation results in accumulation of undegraded ceramide compounds, especially in the brain.
GM1 gangliosidosis is an autosomal recessive LSD caused by deficiency of beta-galactosidase and characterized by neuronal lipidosis and mucopolysaccharidosis-like somatic changes. It presents in infancy with severe psychomotor retardation and a full-blown Hurler phenotype (see MPS further on) including coarse facial features, dysostosis multiplex, corneal clouding, and organomegaly. A cherry-red spot is seen in 50% of patients. Patients with infantile onset usually die before two years of age. Later onset variants have a milder neurological picture (which includes dystonia and ataxia) and milder somatic and skeletal findings.
Neuronal storage |
 MCBs in GM1 gangliosidosis |
The neuropathological changes in infantile and late infantile GM1 gangliosidosis consist of lipid storage in the neuronal soma (neuronal ballooning) and proximal axon. Storage also occurs in sensory and autonomic ganglionic neurons. Sphingolipids have a hydrophobic and a hydrophilic side and tend to form bilayers in aqueous moieties. This property is replicated in the storage products of gangliosidoses, which form membranous cytoplasmic bodies (MCBs) seen on electron microscopy. In early phases of the disease, the brain is large. GM1 ganglioside, normally 20% of brain gangliosides, increases to 80-90%. As the disease advances, neuronal loss and gliosis occur and cerebral atrophy develops. Storage in retinal ganglion cells results in the cherry-red spot. Patients with GM1 gangliosidosis also have skeletal, soft tissue, and visceral changes similar to Hurler syndrome (see mucopolysaccharidoses further on).
The GM2 gangliosidoses are a prime example of the genotypic complexity and phenotypic diversity of LSDs. GM2 accumulation can result from deficiency of hexosaminidase A (Hex A), hexosaminidase B (Hex B), or the GM2 activator, a polypeptide that forms a complex with GM2 ganglioside that is important for its degradation. Hex A catabolizes GM2 ganglioside only. Hex B is multicatalytic and catabolizes GM2 and other substrates with terminal GlcNAC or GalNAC such as oligosaccharides, glycolipids, glycoproteins, and GAGs. Hex A is a heterodimer. It consists of one alpha subunit encoded by the HEXA gene on chromosome15 and one beta subunit encoded by the HEXB gene on chromosome 5. Hex B consists of two beta subunits. Alpha subunit (and Hex A) deficiency causes Tay-Sachs disease (TSD); Beta subunit mutations cause Hex A and Hex B deficiency (Sandhoff disease). TSD is the poster child of LSDs and the first LSD to be described. Patients with classic infantile TSD present in the first year of life and die by five years of age. They have profound psychomotor retardation, myoclonus, hypotonia, and cherry-red spots. Variants caused by milder mutations begin later and have a subacute or chronic course characterized by seizures, ataxia, spasticity, choreoathetosis, and motor neuron disease with little or no cognitive dysfunction.
The neuropathology of infantile TSD is generalized neuronal lipidosis indistinguishable from GM1 gangliosidosis. The brain is larger than normal initially but, by the end of the clinical course, severe neuronal loss and brain atrophy occur. GM2 ganglioside, normally a minor component of total brain gangliosides, increases to 90% of brain gangliosides. The few late onset cases that have been studied show similar but less severe changes throughout the brain, more severe storage in spinal cord neurons and dorsal root ganglia, and more diverse and heterogeneous lysosomal contents.
The neurological manifestations and neuropathology of Sandhoff disease are similar to TSD but Sandhoff disease shows also hepatosplenomegaly and visceral storage of glycolipids oligosaccharides, glycoproteins, and glycolipids. No such visceral storage is seen in TSD. The clinical profile and pathology of activator deficiency are similar to TSD.
METACHROMATIC LEUKODYSTROPHY
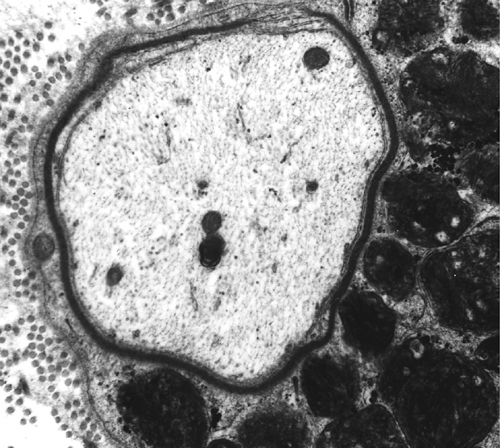
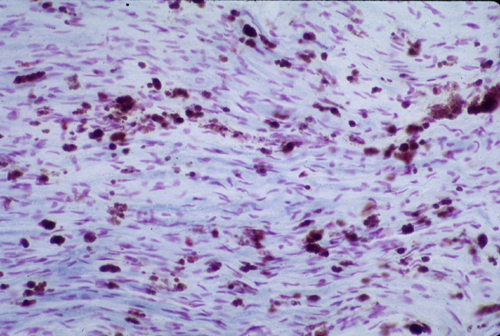
Metachromatic leukodystrophy (MLD) is an autosomal recessive deficiency of arylsulfatase A that results in accumulation of the myelin lipid sulfatide in oligodendrocytes and Schwann cells. In its most common variant, patients are normal up to age one or two years, and then develop progressive peripheral neuropathy, psychomotor retardation, and blindness. Signs of white matter involvement (spasticity, brisk tendon reflexes, extensor plantar responses) are prominent. Less severe variants cause adult onset dementia, psychiatric disorders, and neuropathy.
 MLD:peripheral nerve |
 MLD:loss of myelin |
 MLD: metachromatic deposits |
Lysosomal storage of sulfatides kills oligodendrocytes and Schwann cells. Sulfatides discharged from dying cells are picked up byhistiocytes. The white matter shows diffuse loss of myelin which spares the subcortical fibers. Peripheral nerves show a demyelinative neuropathy. Sulfatides are also asymptomatically stored in a variety of somatic cells such as the epithelium of the gallbladder and renal tubules. Sulfatide deposits stain pink with H&E. With acid cresyl violet, they take on a brown color (brown metachromasia), hence the term metachromatic leukodystrophy.
The biochemical defect of MLD does not involve myelin synthesis, but rather the degradation and recycling of myelin lipids. Myelin is probably formed normally initially. Subsequently, dysfunction and loss of myelin-producing cells cause loss of myelin. Myelination of some tracts begins in utero. The bulk of myelin is produced in the first two years of life. This is when severe MLD becomes clinically apparent. MLD and globoid cell leukodystrophy are LSDs. Other metabolic white matter disorders are presented in a separate page "Leukodystrophies"
GLOBOID CELL LEUKODYSTROPHY (KRABBE'S DISEASE)
About one third of myelin lipid consists of galactosylceramide (galactocerebroside) and its sulfated variant sulfatide. Deficiency of galactosylceramidase (GALC) causes globoid cell leukodystrophy (GCL) or Krabbe's disease. Children with the classic infantile form of GCL appear normal at birth but, in a few months, develop irritability, spasticity, progressive neurological regression, peripheral neuropathy and seizures, and usually die in two or three years, many in a few months. Patients with juvenile-onset GCL have a more protracted course characterized by irritability, neurological regression, spasticity, visual loss, and ataxia, leading eventually to severe disability and death. There are also adult-onset variants with slow progression and a possible normal lifespan. GCL is autosomal recessive.
 Globoid cells |
 Krabbe's disease |
Unlike other LSDs, in which the brain and other organs contain an increased amount of uncleaved substrate, the brain in GCL does not show a large increase of galactosylceramide, probably because myelin, which contains galactosylceramide is lost. Some galactosylceramide in the form of filamentous or tubular inclusions accumulates in brain macrophages which are transformed into large, often multinucleated, PAS-positive, globoid cells. Most of the damage in GCL is caused by accumulation in the white matter of a related metabolite galactosylsphingosine (psychosine), also a substrate of galactosylceramidase, which is toxic to oligodendrocytes. The combined effects of lipid imbalance and toxicity result in severe myelin degeneration. The white matter in classic infantile GCL is devoid of oligodendrocytes and myelin (except for the subcortical fibers), firm because of gliosis, and contains globoid cells, which tend to accumulate around vessels. The cortex is normal and there is no galactocerebroside storage in neurons. There is neuronal loss in the thalamus, cerebellum and brainstem. Peripheral nerves show a demyelinative and axonal neuropathy with accumulation of galactocerebroside in Schwann cells and macrophages. No other organs are affected.
GAUCHER DISEASE
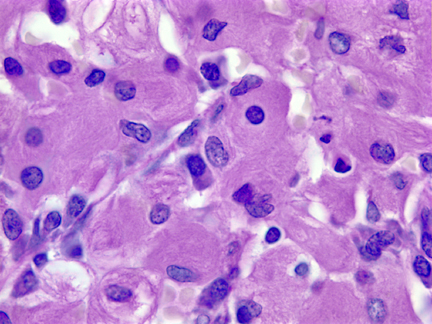
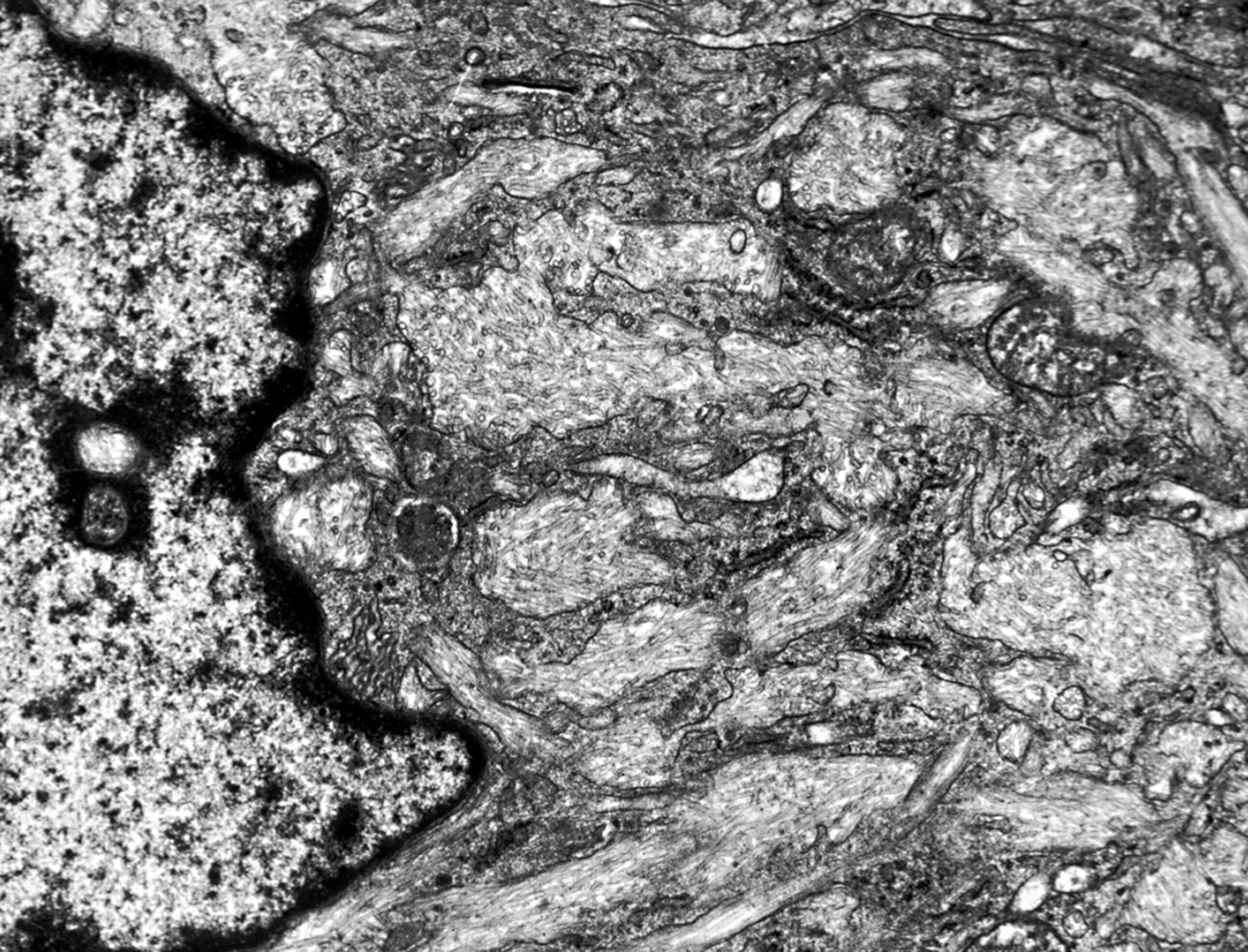
Gaucher disease (GD) is caused by mutations of the GBA1 gene which encodes the enzyme glucocerebrosidase (glucosylceramidase). Deficiency of glucocerebrosidase results in storage of glucocerebroside (glucosylceramide) in monocyte-macrophage cells. Three clinical phenotypes are recognized. The most common is type 1 which is especially prevalent in Ashkenazi Jews. Type 1 GD presents from childhood to early adulthood and causes hepatosplenomegaly, bone disease (osteopenia, focal lytic or sclerotic lesions, osteonecrosis, pathologic fractures, chronic bone pain), anemia and thrombocytopenia due to hypersplenism, and pulmonary interstitial infiltrates. Spinal cord and root compression secondary to bone disease may also develop but there is no storage in the CNS. Type 2 (acute neuronopathic) GD patients have hepatosplenomegaly similar to type 1, but develop also neurological manifestations (stridor, strabismus and other oculomotor abnormalities, swallowing difficulty, opisthotonus, spasticity) which cause their death by 2 to 4 years of age. There is no special ethnic prevalence for type 2 GD. Type 3 (subacute neuronopathic) GD is frequent in Northern Sweden and has hematological and neurological manifestations similar to type 2 but milder and more slowly progressive. GD is the first LSD to be successfully managed by enzyme replacement therapy. Persons with pathogenic GBA1 mutations, including carriers, also have a 20- to 30-fold increased risk for developing Parkinson’s disease (PD), and 7-10% of PD patients have GBA1 mutations.
 Gaucher cells |
 Membrane-bound tubular inclusions in Gaucher cell |
GD is the prototype of storage histiocytosis. Lysosomal storage of glucocerebroside in cells of the monocyte-macrophage system leads to their transformation into Gaucher cells (GC). GCs have a large cytoplasmic mass with a striated appearance that has been likened to "wrinkled tissue paper" or "crumpled silk". On EM, the storage material takes consists of tubular inclusions. GCs are present in the bone marrow, spleen, lymph nodes, hepatic sinusoids, and other organs and tissues in all forms of GD. An increased incidence of cancer including lymphoma, myeloma, and bone tumors has been reported in GD patients. There is no storage in neurons or glial cells. In type 2 and 3 GD, there are numerous GCs in perivascular CNS spaces and rare GCs in brain parenchyma. No part of the CNS is spared but the brainstem and deep nuclei are more severely affected than the cortex and account for most neurological deficits. Along with the presence of GCs, type 2 and 3 GD shows also neuronophagia, neuronal loss, and gliosis. No neuronal storage is seen. Neuronal degeneration and loss have been attributed to the neurotoxic action of glucosyl sphingosine, a by-product of glucocerebroside not normally present in the brain.
MUCOPOLYSACCHARIDOSES (MPS)
Mucopolysaccharides (now called Glycosaminoglycans-GAGs) are synthesized in the Golgi apparatus and secreted and assembled in the extracellular space. They are produced by all cells, and are especially abundant in connective tissues. They are an important component of the matrix of connective tissue, cartilage and bone. For recycling, GAGs are internalized and degraded in a stepwise fashion by lysosomal enzymes. Deficiency of these enzymes causes lysosomal storage of GAGs. There are six clinical groups of MPS caused by deficiencies of ten GAG-cleaving enzymes.
Intracellular storage of GAGs in hepatocytes and other cells causes hepatomegaly, cellular dysfunction, and cell death. The most severe somatic changes in the MPS are due to accumulation of GAGs in matrix due to impaired recycling and to discharge of GAGs from dying mesenchymal cells. Because they are negatively charged, GAGs attract a lot of water that causes their molecules to swell to tremendous volumes. High GAG content of connective tissues affects collagen synthesis and causes increased collagen deposition.
 MPS |
 MPS: thickened cardiac valves |
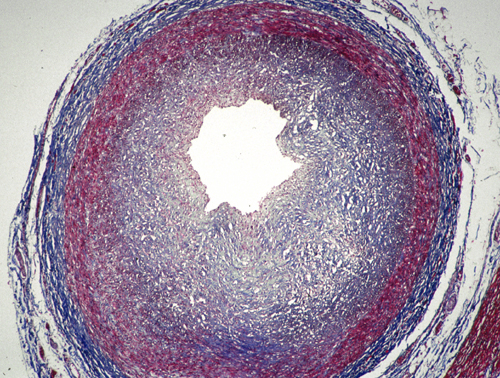 MPS-coronary artery: intimal thickening |
The skin, connective tissues, and cartilage become swollen and distorted. The connective tissue and cutaneous changes cause facial deformity and macroglossia which gave rise to the insensitive term gargoylism. Cardiac valves and chordae tendineae become thickened and stiff. Endocardial and interstitial myocardial fibrosis develops. The intima of coronary arteries may be thickened to the point of occlusion and the aorta develops fibrous intimal plaques without lipid deposition. These changes cause a fatal cardiomyopathy and ischemic heart disease. GAG storage causes joint stiffening and swelling and complex skeletal deformities known as dysostosis multiplex. Storage in corneal fibroblasts causes corneal clouding.
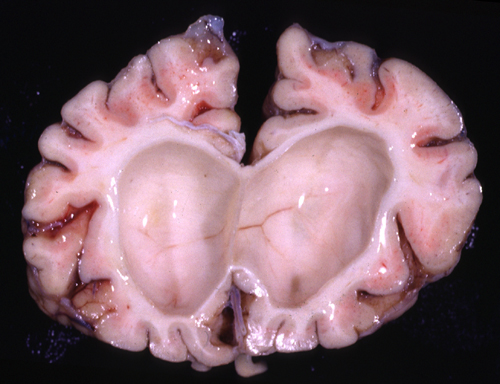 MPS: Hydrocephalus |
 MPS: expanded perivascular spaces |
 MPS: "Zebra bodies" |
GAG deposition in connective tissues of the brain and spinal cord causes thickening of the dura which, along with distortion of vertebrae, results in compression myelopathy. The arachnoid membrane is thickened and the subarachnoid space and parivascular spaaces are expanded.Thickening of the arachnoid membrane impairs CSF flow, causing communicating hydrocephalus. But the most devastating neurological effects of MPS are due to neuronal storage of gangliosides. The mechanism of this storage is poorly understood. It is probably due to inhibition of neuraminidase and other lysosomal enzymes induced by the storage of GAGs. Thus, in addition to the skeletal, cardiovascular and other lesions, many MPS also cause neuronal lipidosis. Gangliosides stored in nerve cells take the form of concentric membranes (membranous cytoplasmic bodies) or stacks of membranes (zebra bodies).
NEURONAL CEROID LIPOFUSCINOSES
 NCL. Curvilinear bodies |
 NCL. Cerebellar atrophy |
Lipofuscin and ceroid are undegradable materials formed in lysosomes by peroxidation of membrane lipids. They take the form of golden-brown, autofluorescent, PAS-positive, and acid-fast granules and are especially prominent in neurons and cardiac myocytes. The neuronal ceroid lipofuscinoses (NCLs) are a group ofautosomal recessive disorders in which similar products accumulate in nerve cells, histiocytes, and many other cells and tissues. They are divided into four main groups, infantile, late infantile, juvenile, and adult NCL and four other less frequent entities. The term Batten disease is used for juvenile NCL, first described about 100 years ago, and is also applied to the entire group of NCLs. The NCLs are the most common causes of neuronal storage disease and, according to some authors, the most common neurodegenerative diseases in children. Pathologically, they show neuronal ballooning due to lysosomal storage of a variety of granular, lamellar, curvilinear, and other products. This storage causes neuronal loss, cortical atrophy, and cerebellar and retinal degeneration resulting in seizures, myoclonus, ataxia, and blindness. The NCLs are inexorably progressive diseases, and most are fatal.
The storage material contains subunit C of mitochondrial ATP synthase and components of sphingolipid associated proteins (saposins). The NCLs are linked to six known CLN genes. Two of these (CLN1 and CLN2) encode lysosomal enzymes, Palmitoyl Protein Thioesterase (PPT) and Tripeptidyl Peptidase 1 respectively, which are involved in the lysosomal degradation of proteins. The products of the other CLN genes are lysosomal and endosomal membrane proteins whose function is unkown. The pathogenesis of NCLs has been unclear. For a long time, no enzyme deficiency could be demonstrated, but now it appears that they belong in the LSDs after all. They are probably caused by deficiencies of lysosomal proteases. Histiocytic storage of ceroid and lipofuscin (sea blue histiocytes) develops also in neoplastic and non-neoplastic blood disorders with accelerated turnover of hematopoietic cells.
NIEMANN-PICK DISEASE TYPE C
Type A and B Niemann-Pick disease are neurovisceral storage diseases caused by deficiency of sphingomyelinase. Niemann-Pick type C (NPC) is an LSD with protean clinical manifestations including neonatal hydrops, neonatal hepatitis, storage histiocytosis and neuronal lipidosis. The material that is stored in lysosomes in NPC is not sphingomyelin but cholesterol. Patients with NPC can import LDL cholesterol into lysosomes and remove the cholesteryl ester generating free cholesterol, but they cannot move free cholesterol to its normal cellular destinations. Thus, cholesterol accumulates in lysosomes. The mutant gene is located on 18q and its product, the NPC1 protein, is a transmembrane protein which acts as "gatekeeper" in the transport of lysosomal cholesterol to its other cellular targets. The "filipin test", which is used for diagnosis of NPC, consists of feeding cultured fibroblasts with LDL cholesterol tagged with the fluorescent dye filipin. The fibroblasts show bright fluorescence due to accumulation of cholesterol. NPC is rare but its study has produced some important insights into intracellular cholesterol homeostasis and trafficking.
Further Reading
- Suzuki K. Globoid Cell Leukodystrophy (Krabbe’s Disease): Update. J Child Neurol 2003;18:595–603. PubMed
- Walkley SU. Pathogenic cascades in lysosomal disease-Why so complex? J Inherit Metab Dis 2009; 32:181-9. PubMed
Updated: April 2023