Prion Diseases (Transmissible Spongiform Encephalopathies)
Transmissible spongiform encephalopathies (TSEs) are caused by abnormal folding of prions (pronounced pree-ons, an acronym for proteinaceous infectious particles). The normal prion protein, which is designated as PrPC, is a 35kD membrane glycoprotein, which is water-soluble and proteinase-sensitive. Abnormal prions, designated as PrPSc, result from a change in the folding pattern of PrPC, which makes it resistant to the action of proteases and causes it to precipitate as insoluble amyloid. This conversion results in neuronal degeneration and loss by an unknown mechanism. PrPCis encoded by the PRNP gene on chromosome 20 and probably plays a role in multiple cellular functions, including cell adhesion, ion channel activity, and neuronal excitability.
Pathogenesis of prion diseases
The unique feature of prion diseases is that they are self-propagating and transmissible. Once PrPSc is generated endogenously or introduced into the body from the environment, it converts normal prions into abnormal ones. This conversion begins with the initial production of a small polymer of misfolded prions, (a seed), perhaps no more than 28 molecules. This seed converts normal adjacent prions into abnormal ones by an unknown mechanism. As more PrPSc polymers are produced, they, in turn, act as seeds, propagating the conversion of normal to abnormal prions. This novel mechanism of disease is also implicated in the propagation of protein misfolding that is involved in Alzheimer's disease, Parkinson's disease, and amyotrophic lateral sclerosis.
The majority of human prion diseases (see table below) are sporadic. About 15% are familial, autosomal dominant. A few are environmentally acquired (iatrogenic and from eating BSE- contaminated meat). In familial prion diseases, the change in PrPC conformation is caused by mutations of the PRNP gene (more than 20 reported), which alter its amino acid sequence. When extrinsic abnormal prions are introduced into the body, they interact with normal indigenous prions and cause them to change their conformation into abnormal. How sporadic prion disease arises, is a mystery. Perhaps the initial seed of PrPSc is caused by somatic mutations or by posttranslational modifications.
The transmissibility of prion diseases has been proven by animal experiments. Natural transmission from animal to animal (especially in sheep) has been known for many years. In a few instances also, prion diseases have been accidentally transmitted from human to human by transplantation of tissues (dural grafts, cornea) or injection of pituitary extracts from patients with prion diseases. In 1996, transmission of bovine spongiform encephalopathy to humans was reported in the UK.
The human prion diseases are:
Creutzfeldt-Jakob disease.
Sporadic-sCJD,
familial-fCJD, iatrogenic-iCJD, and variant-vCJD)(see
below).
Kuru. A now-extinct disease
of New Guinea natives, transmitted by eating
the brains of dead persons who had the disease. Kuru is significant historically as it was the first prion disease to be discovered, and its study established the transmissibility of prion diseases.
Gerstmann-Straüssler-Scheinker syndrome
(GSS). An autosomal dominant slowly
progressive ataxia and dementia, characterized
by widespread PrPSc amyloid plaques
throughout the CNS.
Fatal familial insomnia (FFI).
An autosomal dominant sleep disorder with pathological
lesions in the thalamus.
The most common animal prion diseases are:
Scrapie. An important disease of sheep
that has been known for over 100 years. Sick animals
rub against rocks or other hard surfaces, scraping
their fleeces. The discovery of transmissibility
and other important aspects of the biology of prion
diseases was based on knowledge of scrapie.
Bovine spongiform encephalopathy (BSE)-mad cow disease.
Transmissible mink encephalopathy.
Wasting disease of deer and elk.
Prion diseases have also been reported in several other domesticated and
wild animal species and can cross from one species to another. Experimental
transmission to primates and guinea pigs has played an important role in
elucidating their pathogenesis.
Pathology of prion diseases
During the incubation period and active clinical phase of prion diseases, PrPSc is probably present (and can transmit the disease) in all tissues and body fluids, especially brain, nerves, and skeletal muscle. Pathology develops only in the brain. Initially, intracytoplasmic vacuoles appear in neurons. As the disease progresses, vacuolization becomes more pronounced and the cortical neuropil develops a spongy appearance, hence the term spongiform encephalopathy. Similar spongiform change develops in the cerebellar molecular layer.
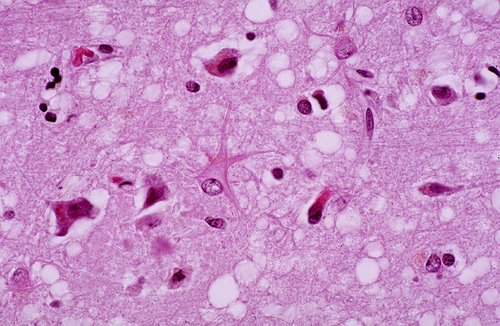 Spongiform encephalopathy |
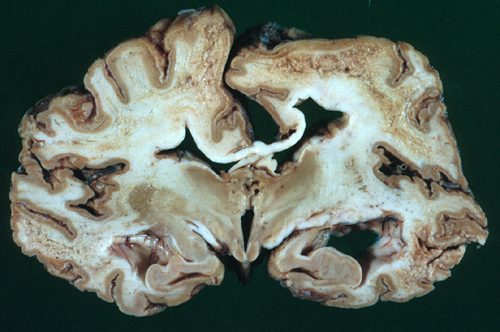 Advanced CJD: severe brain atrophy |
 AdvancedCJD: Cerebellar degeneration |
Advanced cases show neuronal loss, gliosis, and brain atrophy. Cerebellar atrophy is usually severe. Unlike most cerebellar degenerations, there is more pronounced loss of granular neurons than Purkinje cells. There is no inflammation. The CSF is normal. The changes are confined to the gray matter (the primary pathology involves the neuronal body). In some prion diseases, PrPSc precipitates as amyloid plaques.
Creutzfeldt-Jakob Disease (CJD)
CJD is the most common prion disease of humans,
but overall it is rare, affecting one in every million
people. Most cases are sporadic (sCJD).
sCJD affects middle aged or old persons and causes
dementia, myoclonus, ataxia and other neurological
abnormalities. This phenotype, also known as the
Heidenhain variant, is seen in 70% of sCJD. The
other 30% have less typical features. Virtually,
any unexplained or unusual neurological sign or
symptom can be a manifestation of CJD. The EEG shows
characteristic periodic sharp wave complexes (PSWC).
CJD is inexorably progressive and fatal within months
up to 1 to 2 years.
Approximately 15% of CJD cases are familial (fCJD), autosomal dominant. fCJD is clinically indistinguishable from sCJD. Two other inherited prions diseases, GSS and FFI, have distinct clinical and pathological phenotypes. Over 200 cases of variant CJD (vCJD) have been reported, most of them in the UK. It is believed that vCJD is caused by consumption of meat products contaminated by BSE. vCJD patients are young and have prominent psychiatric manifestations and ataxia. The brain shows spongiform encephalopathy and numerous amyloid plaques. Unlike sCJD, prion protein can be detected in lymphoid tissues.
Over 400 cases of iatrogenic CJD (iCJD) have been caused by injection of growth hormone and gonadotrophin extracted from cadaveric pituitaries, contaminated dural grafts, corneal transplants, and contaminated surgical instruments.
LABORATORY DIAGNOSIS OF CJD
EEG: PSWCs
MRI: DWI and FLAIR sequences show high signal abnormalities in the caudate nucleus and putamen or at least 2 cortical regions (temporal, parietal-occipital). Patients with vCJD have high intensity signal in the posterior thalamus (pulvinar).
CSF: Protein and glucose concentration is normal and there is no pleocytosis. The most useful CSF markers of CJD are Protein 14-3-3 and Real-Time Quaking-induced Conversion (RT-QuIC). 14-3-3 is a 30kD protein that plays a role in cell proliferation, differentiation and signal transduction and regulates neurotransmitter synthesis. Protein 14-3-3 is also elevated in patients with encephalitis, cerebral infarction, and other conditions. RT-QuIC replicates prion formation in vitro and is nearly 100% specific for CJD. These tests provide much needed help in the premortem diagnosis of CJD and are especially helpful when the differential diagnosis has been narrowed down and more common neurological conditions such as Alzheimer's disease, stroke, HIV encephalitis, etc have been excluded.
Brain biopsy: A definitive diagnosis can be made by microscopic examination of brain tissue showing the characteristic spongy change. Western blotting of brain tissue extracts following proteinase K digestion reveals electrophoretic patterns that identify different prion strains. Such studies helped establish the identity of PrPSc strain in vCJD and BSE.
DNA sequencing: Mutations of the PRNP gene that cause fCJD and the codon 129 polymorphism that influences sCJD can be detected by sequencing of DNA extracted from blood, brain and other tissues.
The etiology of sCJD is unknown. The story of the vCJD has raised the specter of food contamination as a possible cause, but there is no evidence that sporadic CJD is caused by that mechanism. Somatic mutations of PrP can cause sporadic CJD but most sporadic cases show no such mutation. However, the PrP gene shows a polymorphism at codon 129 that can code for either Methionine or Valine. Most patients with sCJD and iCJD are homozygotes for Methionine. In addition to being a risk factor for developing CJD, this polymorphism affects the physicochemical properties of prions and influences the clinical phenotype and pathology of CJD.
The potential of accidental transmission of a fatal neurological disorder has scared pathologists and other health care workers connected with these diseases. This potential should not be underestimated. However, these fears are exaggerated. The disease is transmitted by invasive methods such as inoculation or injection and not by casual contact such as occurs in the course of routine patient care. An autopsy can be safely done using standard precautions. Fixation of tissues in formalin does not eliminate transmissibility of CJD and other prion diseases. Additional chemical treatment with formic acid is needed.
Before prions were known, sporadic CJD and other prion diseases were thought to be "degenerative" diseases. When it was discovered that they can be transmitted, they were reclassified as infectious diseases. At one point, they were called slow virus diseases. However, they are obviously different from bacterial and viral infections. For one thing, prions contain no nucleic acid, and their chemistry and structure are not like those of bacteria or viruses. They damage neurons directly and do not elicit inflammatory and immune reactions. The familial forms of prion diseases can be regarded as inherited metabolic disorders, similar to Huntington's disease and familial Alzheimer's disease. Understanding the transmitted and sporadic forms is a challenge. An abnormal protein entering the body, crossing the blood-brain barrier, and causing its normal counterparts in neurons to change their folding pattern in a chain reaction that eventually destroys the brain sounds like science fiction. Prions have been compared to the Borg, an alien mechano-anthropoid species in Star Trek, which relentlessly assimilate other species with which they come in contact. Two Nobel prizes have been awarded for research in prion diseases. In 1976, Carleton Gajdusek received the prize for proving that they are transmissible, and in 1997 Stanley Prusiner was given the prize for the prion hypothesis.
Further Reading
- Zerr I .Laboratory Diagnosis of Creutzfeld-Jacob Disease. New Engl J Med 2022;386:1345-50PubMed
- Brown P. Transmissible spongiform encephalopathy in the 21st century: neuroscience for the clinical neurologist. Neurology 2008;70:713-22. PubMed
- Glatzel M, Stoeck K, Seeger H, Lührs T, Aguzzi A. Human prion diseases:molecular and clinical aspects. Arch Neurol 2005;62:545-52. PubMed
- Shiga Y, Miyazawa K, Sato S, et al. Diffusion-weghted MRI abnormalities as an early diagnostic marker for Creutzffeldt-Jakob disease. Neurology 2004;63:443-9. PubMed
- Castellani RJ, Colucci M, Xie Z, at al. Sensitivity of 14-3-3 protein test varies in subtypes of sporadic Creutzfeldt-Jakob disease. Neurology 2004;63:436-42. PubMed
- Zerr I, Kallenberg K, Summers DM, et al. Updated clinical diagnostic criteria for sporadic Creutzfeldt-Jakob disease.Brain. 2009;132(Pt 10):2659-68.PubMed
- Appleby BS, Rhoads DD, Mente K, Cohen ML. A Practical Primer on Prion Pathology. J Neuropathol Exp Neurol. 2018;77(5):346-352.PubMed
Updated: April, 2022