SPECIAL ISCHEMIC LESIONS: PORENCEPHALY, SCHIZENCEPHALY, AND HYDRANENCEPHALY
Fetuses and newborn infants suffer ischemic and hemorrhagic strokes with surprising frequency. Before birth, vascular occlusion may result from embolism from placental vessels, hereditary coagulopathies, maternal cocaine, and other causes, most of which remain unknown. Additional causes of stroke in the perinatal period are birth trauma, vascular spasm from subarachnoid hemorrhage, disseminated intravascular coagulation, and venous thrombosis. Systemic HIE can also cause focal lesions. The pathology of most of these strokes is similar to that of their adult counterparts. This section describes three special ischemic lesions that have their origin in fetal life. These lesions can be regarded as part of a spectrum, the mildest being porencephaly and the most severe hydranencephaly.
 Porencephaly |
Porencephaly (Gr.Poros a passage, ford, pore) was originally defined as a defect that creates a communication between the cerebral ventricles and the subarachnoid space but now it is used to describe any fluid-filled cavity in the fetal or neonatal brain. A thin membrane may separate the cavity from the lateral ventricle or the subarachnoid space. Such membranes rupture in life or during the autopsy. The pathology has its origin in fetal life. Most cases begin as ischemic infarcts that subsequently cavitate, and are usually located in the middle cerebral artery territory. Similar lesions may result from brain necrosis occurring in prenatal infections. The pathogenesis is no different from necrotic lesions that arise in late gestation or in the neonatal period. The main difference is that lesions arising early in gestation heal without gliosis, and the defect is frequently bordered by dysplastic cortex. On the other hand, lesions arising later are associated with gliosis, macrophage reaction, and calcification.
A genetic angiopathy, caused by mutations of COL4A1, the gene that encodes Collagen 4A1 (a component of the vascular basement membrane) causes porencephaly and cerebral hemorrhage in infants and lacunar infarcts, cerebral hemorrhage, and leukoencephalopathy in adults.
 Closed schizencephaly |
 Closed schizencephaly |
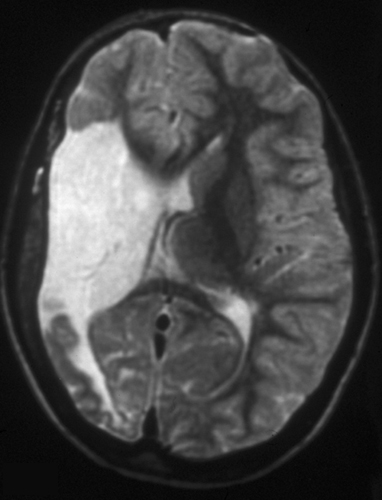 Open schizencephaly |
Schizencephaly (Gr. Schizein, to cleave, slit, split) was originally described by Yakovlev and Wadsworth as “congenital clefts in the cerebral mantle”, extending from the pial surface to the ventricles. The lips of the defects were closed in some cases, forming a seam (closed-lip schizencephaly). In other cases, hydrocephalus caused the lips to separate, creating a gap in the cerebral mantle (open-lip schizencephaly). As in porencephaly, the thin subpial and ependymal membranes along the outer and inner borders of open-lip lesions may rupture, creating a communication between the lateral ventricles and the subarachnoid space. Yakovlev and Wadsworth thought schizencephaly was a malformation caused by failure of development (agenesis) of brain tissue, dating back to the first two months of fetal life. Neuropathologists have dismissed the agenesis hypothesis in favor of an encephaloclastic process and have even discouraged the use of the term schizencephaly. However, the term is used by neuroradiologists, and, thanks to modern imaging, the frequency of schizencephaly appears to be considerably higher than had been previously appreciated.
 Schizencephaly at 20 weeks gestation |
 Schizencephaly occurring in the 3rd trimester |
 Schizencephaly |
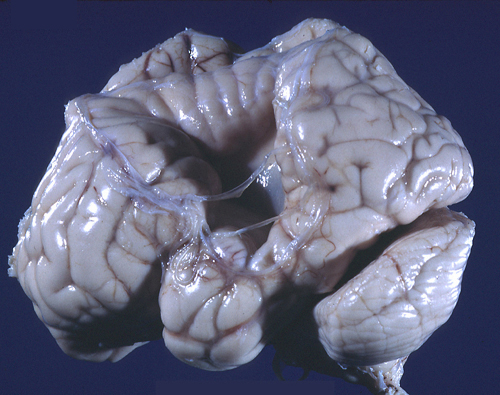 Schizencephaly |
Schizencephaly can be unilateral or bilateral. The location of the lesions on the cerebral convexities suggests ischemic infarcts in the territories of the middle cerebral arteries, as in the cases illustrated on the left. In schizencephaly arising early, no reactive changes are seen. When schizencephaly occurs late in gestation, when the CNS is more mature, the cortex bordering the defects shows gliosis and calcifications, similar to other disruptive lesions. Dysplastic cortex with polymicrogyria is frequently present along the sides of the seam and in the surrounding areas. Some patients have schizencephaly in one hemisphere and polymicrogyria in the same distribution on the opposite hemisphere. The association of schizencephaly with polymicrogyria is so frequent that the two entities are now considered to be part of a spectrum. A few familial cases of schizencephaly have been described.
The neurodevelopmental outcome correlates with the severity of pathology. Unilateral closed-lip schizencephaly has the mildest clinical picture, and bilateral open-lip schizencephaly has the most severe clinical picture. The most prominent manifestations are motor deficits and seizures. Language function is less severely affected. Schizencephaly may be asymptomatic during the first year of life, and 17% of cases have minimal or no symptoms.
 Hydranencephaly |
 Transillumination |
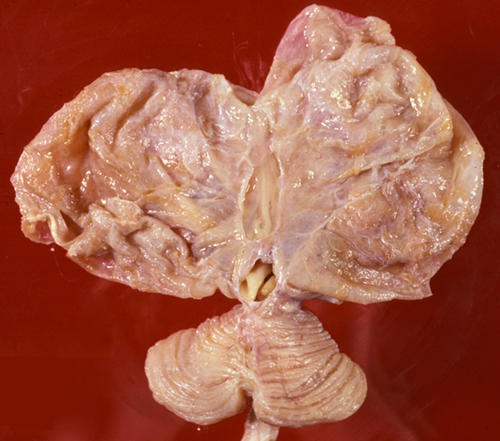 Severe brain atrophy following HIE |
In hydranencephaly, the cerebral hemispheres are replaced by a thin-walled, fluid-filled cyst. The aqueduct is usually atretic, and increased fluid pressure causes the cyst (and the head) to enlarge. There is variable preservation of the inferior frontal, temporal, and occipital lobes, and of the basal ganglia and diencephalon. The brainstem and cerebellum are usually preserved. Hydranencephaly is not a malformation, but a disruption caused probably by ischemia in utero in the territories of the carotid arteries. Some cases of perinatal HIE come close to hydranencephaly. Most children with hydranencephaly die at birth. Some may appear normal initially because the brainstem is intact. Hydranencephaly may be difficult to distinguish by CT or MRI from severe congenital hydrocephalus. The empty cranial cavity transilluminates.
Further Reading
- van der Knaap MS, Smit LM, Barkhof F, et al Neonatal porencephaly and adult stroke related to mutations in collagen IV A1. Ann Neurol. 2006;59:504-11.PubMed
- Merello E, Swanson E, De Marco P et al. No major role for the EMX2 gene in schizencephaly. Am J Med Genet A. 2008;146A:1142-50. PubMed
- Pavone P, Praticò AD, Vitaliti G, et al. Hydranencephaly: cerebrospinal fluid instead of cerebral mantles. Ital J Pediatr. 2014; 40: 79. PubMed
Updated: January, 2018