DENERVATION ATROPHY
 Denervation atrophy |
Diseases that affect the lower motor neuron at any point cause myofiber atrophy. The motor neuron exerts a trophic influence on muscle. This influence is mediated by induced contractions and by chemical substances (trophic factors) released at the synapse, which influence protein synthesis in muscle. Myofibers that lose their innervation become angular and shrink. They lose 80% to 90% of their mass within a few months. At an extreme stage of atrophy, virtually all sarcoplasm is lost and the myofiber is reduced to a cluster of nuclei. In the process of denervation, there is loss and disarray of myofilaments but no myonecrosis occurs. Myofiber atrophy can be best appreciated in cross sections.
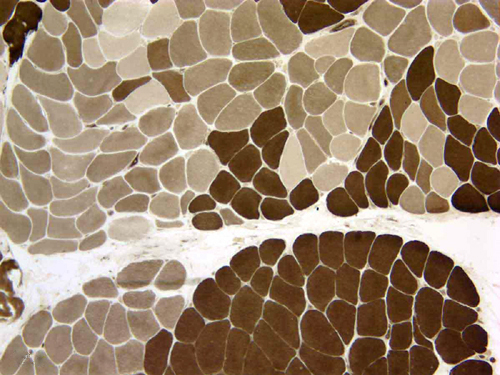 Fiber type grouping |
 Group atrophy |
 Target fibers |
At an early stage, denervation causes atrophy of isolated myofibers, which are scattered in a random fashion. In chronic denervating processes such as chronic neuropathy and motor neuron disease, remaining healthy axons sprout and synapse with denervated fibers (collateral reinnervation). As a result of the combined denervation and reinnervation, motor units enlarge, and their fibers, instead of being scattered, come to lie adjacent to one another. In histochemical stains, such motor units appear as groups of myofibers of the same histochemical type (fiber type grouping). When ultimately these motor units lose their innervation and there are no healthy axons left to connect with them, all their fibers shrink together (group atrophy).
Reinnervation often causes a change in myofibers, the target fiber, characterized by absence of oxidative enzyme activity in the center, surrounded by a rim of more intense than normal activity. Target fibers may be confused with central cores, which occur in a congenital myopathy-central core myopathy.
In the EMG, large motor units appear as polyphasic or giant motor unit potentials. Denervated muscle is overexcitable. Spontaneous discharges from individual myofibers are picked up by the EMG as fibrillations. Spontaneous firing of an entire motor unit causes cotraction of a small group of myofibers that appears as a ripple on the surface of the muscle (fasciculation). Fasciculations of the tongue are an important sign of motor neuron disease. Since denervation does not cause myonecrosis, there is no elevation of CK.
Denervation atrophy is caused by peripheral neuropathies and motor neuron diseases. The most common motor neuron disease in adults is amyotrophic lateral sclerosis. In children, it is the autosomal recessive spinal muscular atrophy and its variants (see Chapter 9-Neurodegeneration). Lower motor neuron damage may also be caused by enteroviruses which include the poliomyelitis virus and some arthropod borne viruses, especially West Nile Virus.
 Arthrogryposis |
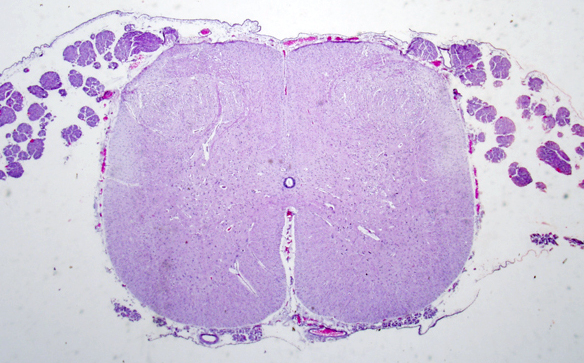 Spinal cord in arthrogryposis |
 Loss of muscle in AMC |
ARTHROGRYPOSIS (claw joints) MULTIPLEX CONGENITA (AMC) refers to a condition in which multiple joint contractures are present at birth. Contractures (ankylosis) are the result of fetal immobility (akinesia, hypokinesia). Immobility has many causes. Mild AMC is caused by oligohydramnios (fetal constriction syndrome). A few AMC cases are due to primary muscle disease (congenital myopathy, congenital muscular dystrophy) and even fewer to congenital neuropathies. Many cases are associated with CNS abnormalities. These include diverse developmental malformations and acquired brain lesions such as hydrocephalus, hydranencephaly, polymicrogyria, microcephaly, and CNS pathology associated with trisomy 18 and other chromosomal abnormalities. The common denominator of these lesions is impairment of fetal mobility because of upper motor neuron dysfunction. A subset of AMC cases are caused by loss of lower motor neurons in the spinal cord and brainstem leading to weakness and denervation atrophy of muscle. The anterior horns of the spinal cord, in these cases, are small and the neuronal groups are disorganized. In some of these cases, loss of lower motor neurons is a component of broader CNS pathology and may coexist with upper motor neuron lesions. In others, loss of lower motor neurons is the only pathology. These cases suggest a burned out poliomyelitis-like process and could conceivably represent the end result of a fetal viral infection. Others may represent an advanced stage of a neurodegenerative processes that evolves during intrauterine life. Muscles, in such cases, are severely wasted, sometimes replaced by fat, suggesting that innervation was lost during the phase of muscle development.
In addition to underlying CNS and spinal cord pathology, in many cases, contractures are a component of numerous genetic disorders with defined genotypes and phenotypes. The term Pena-Shokeir syndrome (PSS) refers to cases of AMC that also have craniofacial abnormalities, polyhydramnios, short umbilical cord, and pulmonary hypoplasia. The PSS is not a cohesive syndrome but rather a phenotype with diverse underlying etiology, pathology, and genetics. Most PSS cases are fatal and many are genetic. A large group of AMC has no CNS or other abnormalities. This group is called amyoplasia and represents the classic arthrogryposis of the orthopedic literature (see further on). Most of these cases are sporadic.
The clinical course of AMC is static. A rare exception is AMC that occurs in cases of spinal muscular atrophy (SMA) of early intrauterine onset. These patients may have mild contractures and their clinical picture is dominated by progressive hypotonia and weakness. Prognosis, in AMC, depends on the severity and extent of contractures and coexisting abnormalities. Some cases of AMC are lethal because of associated CNS and other pathology, especially pulmonary hypoplasia.
 Type 1 fiber predominance in AMC |
The muscle biopsy in AMC is diagnostic in the rare myopathies that cause AMC and in cases of denervation atrophy (provided an affected muscle is sampled). Denervation changes include classic individual or group myofiber atrophy. A significant proportion of biopsies show type 1 fiber predominance, which is probably the end result of denervation and collateral reinnervation. In the rest of AMC cases the muscle biopsy shows nonspecific findings such as myofiber atrophy and replacement of lost muscle with fibroadipose tissue. In some cases, muscle wastes so that only fibroadipose tissue remains. This probably gave rise to the notion of amyoplasia (i.e., agenesis or absence of development of muscle). The term amyoplasia is used in literature to identify a clinical subset of AMC, not as a pathological diagnosis. There is no evidence of agenesis of muscle in AMC cases with detailed pathological studies.
 Type 2 atrophy |
Immobilization or disuse of muscle causes atrophy of type 2 fibers. Type 2 fiber atrophy is also seen with prolonged use of corticosteroids (steroid myopathy) and in polymyalgia rheumatica. Atrophy of type 2 fibers is also seen in sarcopenia ( from the Greek sarx:flesh and penia:paucity), which is characterized by loss of muscle strength and mass. Sarcopenia is mainly associated with old age but can also be secondary to cancer and other systemic diseases, anorexia, diabetes and conditions that cause breakdown of muscle. In sarcopenia there is gross atrophy of muscle, atrophy and loss of myofibers, interstitial fibrosis and replacement of lost muscle by adipose tissue.
Further Reading
- Banker BQ. Arthrogryposis multiplex congenita: spectrum of pathologic changes. Hum Pathol 1986;17:656-72. PubMed
- Hall JG, Reed SD, Driscoll EP. Part I. Amyoplasia: A Common, Sporadic, Condition with Congenital Contractures. Amer J Med Genet 1983;15:571-90. PubMed
Updated: March, 2023