MOTOR NEURON DISEASES
Amyotrophic Lateral Sclerosis (ALS) is a fatal degenerative disorder of upper and lower motor neurons. Lower motor neuron loss causes initially increased electrical excitability leading to fasciculations, and later muscle weakness and atrophy; upper motor neuron involvement causes spasticity, clonus, hyperactive tendon reflexes, and Babinski signs. Most ALS patients present with spinal symptoms (spinal ALS). About one third present with brainstem symptoms (dysarthria, difficulty swallowing) followed by extremity weakness. This variant is called progressive bulbar palsy or bulbar ALS. Bulbar signs also develop in spinal ALS with progression of the disease. Primary lateral sclerosis is an ALS variant that affects upper motor neurons only. Some ALS patients have only lower motor neuron involvement (progressive muscular atrophy). Ten percent of cases present with cognitive and language impairment, behavioral abnormalities and eventually dementia. These cases fall in the spectrum of ALS-frontotemporal dementia (ALS-FTD)(see below). Signs of FTD can also be detected in a much larger proportion of ALS patients. ALS is relentlessly progressive. The majority of patients die, usually from respiratory paralysis, within 3-5 years from the onset of symptoms, sooner in bulbar ALS. There is no specific diagnostic test for ALS. The diagnosis is based on a combination of clinical and electrodiagnostic findings in conjunction with other laboratory tests to rule out diseases that mimic ALS.
Most ALS cases are sporadic and usually affect patients in their 50s. About 10% are familial, mostly autosomal dominant, and have a younger onset. Sporadic cases affect men twice as frequently as women. Familial ALS (FALS) affects men and women equally. Several gene mutations have been discovered in FALS. These mutations affect protein stability, RNA biology, cytoskeletal maintenance, and other functions. Mutations of the same genes are also found in a significant proportion of sporadic ALS cases. The most common gene mutation associated with familial and some sporadic ALS cases is hexanucleotide (GGGGCC) repeat expansion in a noncoding region of C9ORF72 gene on 9p21. This mutation is associated with FTD and TDP-43 inclusions. The second most common cause of FALS is mutation of a gene on chromosome 21q that encodes Cu/Zn superoxide dismutase (SOD1). This enzyme protects cells from toxic oxygen radicals. However, the disease in these cases is not due to loss of antioxidant properties of SOD1 but to a toxic action of the mutated enzyme. This toxicity may have to do with decreased Zn binding by the mutant SOD1. Glutamate toxicity and oxidative stress have been implicated in the pathogenesis of sporadic ALS, and anti-glutamate therapy with Riluzole and anti-oxidant therapy with edaravone prolong survival in some patients.
About 4% of FALS cases are caused by mutations of the RNA processing gene TARDBP. Degenerating motor neurons in FALS due to TARDBP mutations contain ubiquitin-positive neuronal and glial inclusions, which are immunoreactive for TDP-43. TDP-43 is normally a nuclear protein. In FALS due to TARDBP mutations, it is translocated into the cytoplasm, cleaved, ubiquitinated, and phosphorylated. In this form, TDP-43 is presumably toxic. TDP-43 inclusions in TARDBP related ALS are also present in the cerebral cortex, striatum, substantia nigra, and other locations. Identical inclusions are also seen in most cases of TDP-43
fronto-temporal lobar degeneration. Taken together with the presence of dementia in some ALS cases, these observations suggest that this form of ALS is not just a motor neuron disease but a multisystem neurodegenerative disorder, and is part of the spectrum of TDP-43 proteinopathy, which includes ALS and FTD. TDP-43 deposits are not seen in SOD1 mutations. About 4% of FALS cases are caused by mutations of FUS, which also encodes an RNA processing protein.
 Loss of motor neurons in the hypoglossal nuclei |
 Denervation atrophy |
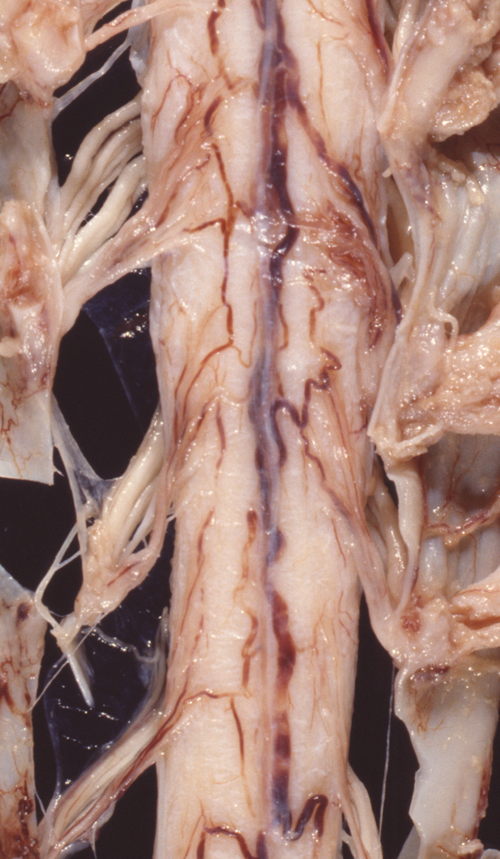 Atrophy of anterior roots |
 Degeneration of the corticospinal tracts |
The pathology of ALS is degeneration and loss of motor neurons in the anterior horns and motor nuclei of brain stem, associated with gliosis, microglial activation, and cytoplasmic deposits of TDP-43, ubiquitin, and SOD1. Eosinophilic cytoplasmic inclusions-Bunina bodies-are present in anterior horn neurons in many cases. There is atrophy of anterior roots. Because of loss of lower motor neurons, muscles undergo denervation atrophy. There is also degeneration of upper motor neurons. As the name of the disease indicates, this is most evident in the lateral corticospinal tracts, which lose axons and myelin and become gliotic. The anterior corticospinal tracts are affected in a similar fashion. Involvement of the internal capsule and motor cortex is usually mild or inapparent, but in severe cases there is loss of upper motor neurons (Betz cells). Degeneration may also infrequently involve sensory tracts. There is also neuronal loss with TDP-43 deposits in parts of the cortex associated with speech and cognition. The latter is more severe in bulbar ALS.
Spinal Spinal Muscular atrophy (SMA) is a genetic disorder that causes degeneration and loss of spinal and brain stem motor neurons (lower motor neurons) resulting in progressive muscle weakness. It is the most common fatal genetic disorder in children and the second most common autosomal recessive disease after cystic fibrosis. SMA is caused by deficiency of a protein encoded by the Survival Motor Neuron gene (SMN) on chromosome 5q11-q13.Normal persons have two forms of the SMN gene, the telomeric (SMN1) and centromeric (SMN2), arranged in tandem. SMN1 produces a functional protein. SMN2 is identical to SMN1 except for a base pair variation that excludes exon 7 from transcription, leading to degradation of its product. However, this exclusion is not complete; SMN2 produces a small amount of functional SMN protein. Most SMA cases are caused by homozygous deletion of SMN1. Individuals with loss of functioning SMN1 gene have multiple copies of SMN2 that produce a small amount of SMN protein. Variation in SMN2 copy number accounts for the variability of SMA phenotype-the more copies of SMN2 the milder the phenotype. SMN protein is important in RNA processing in all cells and appears to have a critical but unknown function in survival of motor neurons. Rare SMA cases are caused by intragenic SMN mutations and mutations of other genes. New genetic therapies for SMA aim at modifying the structure of the SMN2 mRNA resulting in increased production of SMN protein.
| SMA type | Age at onset | Motor Function | Age at death |
|---|---|---|---|
SMA 0 |
Prenatal |
Unable to breathe, arthrogryposis |
<1 month |
| SMA 1 Werdnig-Hoffmann disease | 0-6 months | Unable to sit | <2 years |
| SMA 2 | 6-18 months | Unable to stand | >2 years |
| SMA 3 Kugelberg-Welander disease | 18 months-3 years | Able to stand and walk | Adult |
| SMA 4 | Adult | Able to stand and walk | Adult |
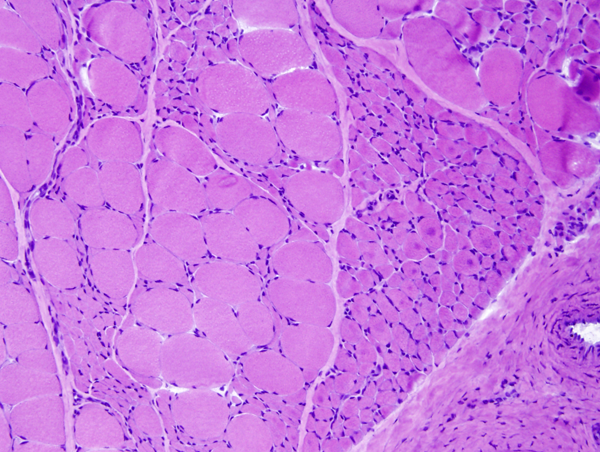 SMA muscle |
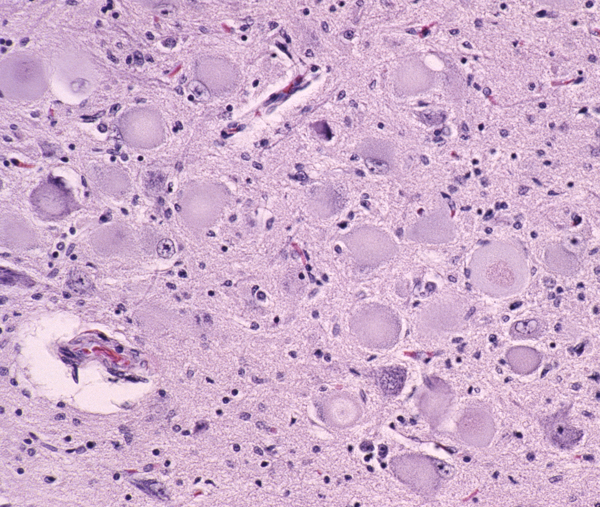 SMA 1 anterior horn |
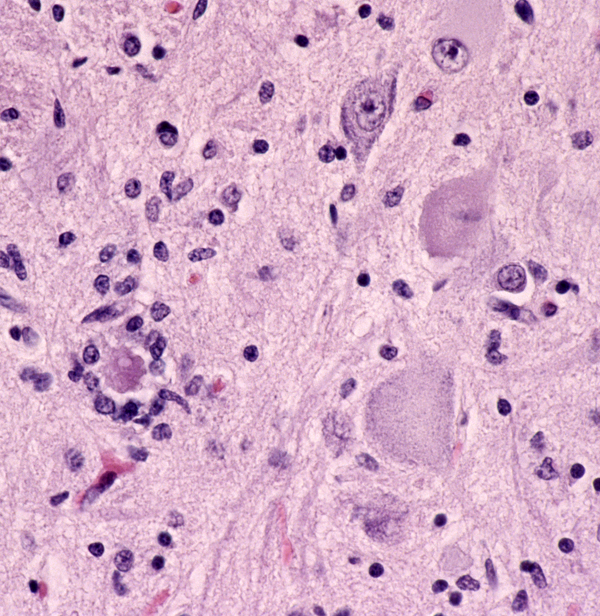 SMA 1 anterior horn |
 SMA anterior horn |
At one end of the spectrum, SMA 0, are cases that have a prenatal onset, paralysis of facial and extraocular muscles, and a very short survival. Arthrogryposis (joint contractures) is seen in some of these cases. At the other end, there are cases that begin in young adult life and have a slow progression. The best known form, SMA 1 (Infantile or Acute SMA; Werdning-Hoffmann disease), begins in infancy and is usualy fatal within two years. Loss of lower motor neurons causes denervation atrophy of muscle, manifested by severe hypotonia, weakness, and inability to breathe. Degenerating brain stem and spinal motor neurons undergo chromatoysis and die. Activated microglial cells often surround and ingest degenerated neurons (neuronophagia). In advanced cases, there is gliosis. In some SMA 1 cases, other neuronal groups besides lower motor neurons (cerebral cortex, basal ganglia, thalamus, brain stem, cerebellum, sensory neurons) are affected. SMA1 must be distinguished from other causes of neonatal hypotonia which include CNS malformations, metabolic diseases, infections, and congenital myopathies. Fasciculations of the tongue, which are a prominent feature of SMA, are helpful in making this distinction. SMA 3 (Juvenile Spinal Muscular Atrophy; Kugelberg-Welander disease) begins in early childhood and has a slow progression, compatible with long survival. It tends to cause proximal weakness and may be confused with myopathy. Patients can walk initially but usually lose this ability later. The difference in severity between the various SMN types has to do with the number of SMN2 copies. So, SMA 0 patients have 1 copy, SMA 4 have 4 copies, and intermediate forms have 2-4 copies.
The changes of denervation are obvious in the muscle biopsy. Today, the diagnosis of SMA is usually made by DNA analysis. Most cases of SMA can be diagnosed with a DNA test that detects deletion of exon 7 from the telomeric copy of the SMN gene. There is no correlation between the type of mutation and the phenotype.
Besides autosomal recessive SMA caused by SMN mutations, there are other, less frequent autosomal recessive, dominant, and X-linked SMA syndromes. There are also rare progressive, autosomal recessive upper motor neuron degenerations (Infantile–Onset Ascending Hereditary Spastic Paralysis, Juvenile Primary Lateral Sclerosis, and Juvenile ALS) which begin during childhood and present with spasticity and weakness. An adult onset, X-linked spinal and bulbar muscular atrophy (Kennedy's disease) is associated with CAG trinucleotide repeats.
Further Reading
- Geser F, Brandmeis MS, Kwong LK, et al. Evidence of Multisystem Disorder in Whole-Brain Map of Pathological TDP-43 in Amyotrophic Lateral Sclerosis. Arch Neurol 2008; 65:636-41. PubMed
- Kiernan MC, Cheah BC, Turner MR, et al. Amyotrophic lateral sclerosis. Lancet 2011;377:942-55. PubMed
- Ince PG. Highley JR, Kirby J, Wharton SB, Takahashi H, Strong MJ, Shaw PJ. Molecular pathology and genetic advances in amyotrophic lateral sclerosis: an emerging molecular pathway and the significance of glial pathology. Acta Neuropathol 2011;122:657–671. PubMed
- Kolb SJ, Kissel JT. Spinal Muscular Atrophy. A Timely Review. Arch Neurol. 2011;68:979-984. PubMed.
- Harding BN, Kariya S, Monani UR, et al. Spectrum of Neuropathophysiology in Spinal Muscular Atrophy Type I. J Neuropathol Exp Neurol 2015;74:15-24.PubMed.
- Brown RH, Al-Chalabi A.Amyotrophic Lateral Sclerosis. New Engl J Med 2017;377:162-172. PubMed.
Updated: April, 2021