This section deals with the principles of lysosomal, peroxisomal, mitochondrial, and amino acid disorders, and highlights some important entities in these groups. There are many more inherited metabolic diseases that are beyond the scope of this web site. Many neurodegenerative diseases and muscle diseases are inherited metabolic disorders, the molecular and biochemical pathways of which we are now beginning to understand.
The diseases covered in this section are, for the most part, childhood disorders. In most of them, patients are normal at birth and have progressive neurological deterioration beginning at some later time. In some of them, the disease is manifested in adulthood. The clinical phenotype depends on the type and severity of the biochemical defect, i.e., what functions are lost and whether the loss is total or partial, and on structural-functional reserves, i.e., what resources are available to replace or cope with the loss. Most inherited metabolic disorders are autosomal recessive.
LYSOSOMAL STORAGE DISORDERS-GENERAL PRINCIPLES
The lysosomal storage disorders (LSDs) are due to deficiencies of lysosomal enzymes caused by mutations of genes that encode the enzyme proteins and related cofactors. Lysosomal enzymes degrade most biomolecules. The products of this degradation are recycled. This process is crucial for the health and growth of cells and tissues. LSDs result in accumulation (storage) of undegraded products in lysosomes. This causes enlargement of cells (ballooning), cellular dysfunction, and cell death. On electron microscopic examination, the stored products are membrane-bound because they are contained within lysosomes.
LSDs are rare. The most common among them are the mucopolysaccharidoses (MPS), which affect one in every 100,000 to 200,000 liveborn infants. The single most common LSD is Gaucher disease. Most LSDs are autosomal recessive. A few are X-linked. Patients are normal at birth. Manifestations of neurological disease begin in infancy or childhood. Initially, there is delay and then arrest of psychomotor development, neurological regression, blindness, and seizures. Inexorable progression leads to a vegetative state.
CLINICAL MANIFESTATIONS AND PATHOLOGY OF LSDs
The clinical manifestations of LSDs depend on which cells and tissues use the deficient enzyme and when is the period of its peak demand. For instance, neurons recycle large amounts of certain gangliosides which are components of their membranes and synapses. Enzymes of ganglioside degradation are highly expressed in brain tissue and are in great need at all times but especially in the first few years of life when axons elongate, dendrites branch, and synapses develop. Deficiency of these enzymes causes neuronal storage of gangliosides. Other gangliosides are components of myelin and their storage causes white matter disease.
LSDs have diverse clinical manifestations. Some of them share certain clinical and pathological features, on the basis of which four basic clinical-pathological phenotypes can be defined: neuronal lipidosis, leukodystrophy, mucopolysaccharidosis, and storage histiocytosis. The most prevalent phenotype is neuronal lipidosis. A few LSDs have distinct clinical features.
CLINICOPATHLOGICAL LSD PHENOTYPES
| PHENOTYPE | PATHOLOGY | CLINICAL FINDINGS | LSDs |
| NEURONAL LIPIDOSIS | Storage in the neuronal body and processes | Neurological regression, seizures,blindness | Gangliosidoses, mucopolysaccharidoses, neuronal ceroid lipofuscinoses |
| LEUKODYSTROPHY | Storage in oligodendrocytes and Schwann cells | Neurological regression, spasticity, peripheral neuropathy | Gangliosidoses (metachromatic leukodystrophy, Krabbe's disease) |
| MUCOPOLYSACCHARIDOSIS | Storage in extraneural tissues | Visceromegaly, soft tissue swelling, skeletal dysplasia, heart disease | Mucopolysaccharidoses, glycoproteinoses, GM1 gangliosidosis |
| STORAGE HISTIOCYTOSIS | Storage in histiocytes | Hepatosplenomegaly, hematopoietic abnormalities | Gangliosidoses (Gaucher disease, Niemann-Pick disease |
The pathology, in the neuronal lipidosis phenotype, involves the gray matter. Storage causes neuronal ballooning and torpedo-like swellings of proximal axons and dendrites.
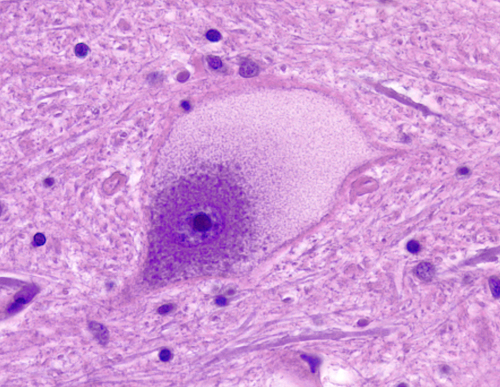 Neuronal storage |
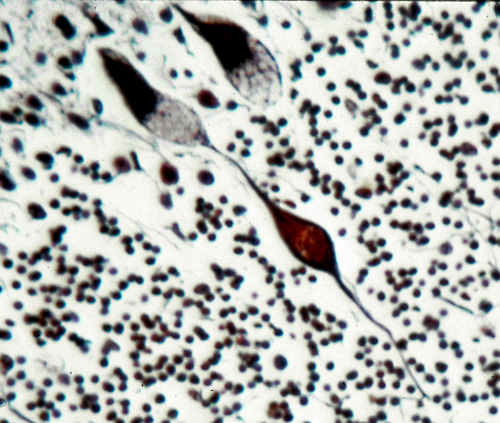 Axonal storage |
 Tay-Sachs disease |
This process leads to loss of neurons and their axons resulting in cortical atrophy and white matter degeneration. The white matter pathology may resemble leukodystrophy (see further on) but the changes are secondary and myelin is not primarily affected. Storage in retinal ganglion cells causes blindness. Other cells and organs that do not process large amounts of gangliosides are either normal or show mild storage without cell damage. The prototype of the neuronal lipidosis phenotype is Tay-Sachs disease, a form of GM2 gangliosisosis prevalent among Ashkenazi Jews, described more than 100 years ago. Some LSDs impair enzymes that are important for turnover of myelin lipids and damage myelin-producing cells. This results in loss of myelin (leukodystrophy) manifested by neurological deterioration and spasticity. The mucopolysaccharidoses and glycoproteinoses affect neurons but have also severe skeletal and visceral manifestations which constitute the mucopolysaccharidosis phenotype. In general, the cells that are most severely affected by LSDs are neurons, because they process large amounts of gangliosides and once lost they cannot be replaced, and histiocytes, because their main function is lysosomal degradation, and they decompensate if their lysosomal enzymes are deficient. Several LSDs show lipid storage in histiocytes throughout the lymphoid and hematopoietic tissues. Such storage histiocytosis causes hepatosplenomegaly, bone marrow depression, bone damage and other manifestations. In some LSDs, notably Gaucher disease, storage histiocytosis is the main abnormality.
The severity of LSDs (which influences the time of onset and the pace of the disease) depends on the residual enzyme activity. The lysosomal enzyme system has a lot of redundancy. Twenty percent of normal enzyme activity is usually adequate to carry out cellular function. Consequently, heterozygotes (carriers of LSDs) whose enzyme activity is 50% of normal are clinically unaffected. Symptoms develop when residual enzyme activity falls below a threshold of 15% to 20%. LDS show allelic variation (different mutations of the same gene). Mutations that leave no residual enzyme activity cause severe, early-onset illness. Milder mutations cause insidious, late- or adult-onset illness.
LSD CLASSIFICATION
The classification of the LSDs is based either on the deficient enzyme or on the chemical composition of the storage material. Eponymic and clinical terms supplement the biochemical nomenclature. In terms of the storage material, LSDs can be divided into three large groups, the sphingolipidoses, mucopolysaccharidoses, and glycoproteinoses and several other individual entities. Sphingolipids consist of a backbone of ceramide with attached oligosaccharide side chains. They are major constituents of cell membranes. Gangliosides have sialic acid side chains and are especially abundant in neuronal membranes. Galactosylceramide and sulfatide are myelin lipids. Glycosaminoglycans (mucopolysaccharides) are long unbranched molecules of repeating disaccharides. They are attached to core proteins forming proteoglycans. They are produced by most cells and are found mainly on the surface of cells and in the extracellular matrix. They are primarily structural molecules. Glycoproteins are also stuctural molecules, components of mucinous secretions, and have a variety of other functions.
Most LSDs are caused by deficiencies of enzymes that degrade carbohydrate side chains and their storage materials are carbohydrates or other glycocompounds. The table below gives a simplified classification of the most common LSDs.
THE MOST COMMON LSDs
| LSD | DEFICIENT ENZYME | PHENOTYPE |
| SPHINGOLIPIDOSES | ||
| GM1 gangliosidosis | b-galactosidase | neuronal
lipidosis mucopolysaccharidosis |
| GM2 gangliosidosis (Tay-Sachs disease, Sandhoff diseasse) |
hexosaminidase A, hexosaminidase B |
neuronal lipidosis |
| Niemann-Pick Disease | sphingomyelinase | neuronal lipidosis storage histiocytosis |
| Globoid cell leukodystrophy (Krabbe dis) |
galactocerebrosidase | leukodystrophy |
| Metachromatic leukodystrophy | arylsulfatase A | leukodystrophy |
| Gaucher disease | glucocerebrosidase | storage histiocytosis |
| MUCOPOLYSACCHARIDOSES | glycosaminoglycan cleaving enzymes | mucopolysacccharidosis |
| GLYCOPROTEINOSES | glycoprotein cleaving enzymes | mucopolysacccharidosis |
| GLYCOGENOSIS TYPE II (POMPE DISEASE) | a-glucosidase | skeletal and cardiac myopathy |
| NEURONAL CEROID LIPOFUSCINOSES | lysosomal proteases | neuronal lipidosis |
LABORATORY DIAGNOSIS OF LSDs
The gold standard for diagnosis of LSDs is enzyme assay. For most LSDs, this can be performed on leukocytes with fast turnaround. It is important to narrow down the differential diagnosis to help decide which assay to order. Cultured fibroblasts are required in a few LSDs. Cultured amniocytes or chorionic villus cells may be used for prenatal diagnosis. Biochemical determination of storage products is cumbersome, but has some applications. For instance, demonstration of GAGs in urine is a useful screening test for GAG storage. Storage of abnormal products can be detected by light and electron microscopy. In addition to neurons, gangliosides and ceroid-lipofuscin are stored in somatic cells and may be detected by nerve, muscle, skin, conjunctival, and other biopsies. Tissue diagnosis (detection of specific storage materials by electron microscopy) is still the standard for some NCLs because no other laboratory tests are available. The gene mutations of LSDs can be detected by DNA analysis. Mutation analysis is used mainly for carrier detection.