DEMYELINATIVE DISEASES
Demyelinative diseases of the central nervous system are characterized by loss of myelin with variable loss of axons. In contrast, infarcts, contusions, encephalitis, and other conditions destroy myelin and axons equally. The main demyelinative disease of the CNS is multiple sclerosis (MS) and its variants. Its counterpart in the peripheral nervous system is inflammatory demyelinative polyradiculoneuropathy (Guillain-Barré syndrome-GBS) and its chronic variants. MS and GBS are autoimmune inflammatory diseases. There are also virus-induced demyelinative diseases, such as progressive multifocal leukoencephalopathy. Demyelinative diseases should be distinguished from leukodystrophies, which are inherited metabolic disorders of myelin lipids and proteins.
MS affects one in every 500 persons, women three times as frequently as men. It is more common in young adults and causes a variety of neurological deficits (visual loss, paralysis, sensory loss, ataxia, brainstem signs, psychiatric disorders, dementia). Many MS cases evolve over a long period (20-30 years) with remissions and exacerbations. Some cases have an acute, fulminant, even fatal course, and others go into a relentlessly progressive phase after a period of remissions and exacerbations.
PATHOLOGY OF MS
The pathology is characterized by multifocal lesions, the MS plaques. The usual evolution of the MS plaque is as follows: in the acute phase (active plaque), activated mononuclear cells, including lymphocytes, microglia, and macrophages destroy myelin and, to a variable degree, oligodendrocytes. Myelin debris are picked up by macrophages and degraded. At an early stage, macrophages contain myelin fragments; later, they contain proteins and lipids from chemical degradation of myelin. This process takes a few weeks. With time, gliosis develops, and plaques reach a burned-out stage consisting of demyelinated axons traversing glial scar tissue (inactive plaque). Remaining oligodendrocytes attempt to make new myelin. If the inflammatory process is arrested at an early phase, plaques are partially remyelinated (shadow plaque). In more advanced lesions, remyelination is ineffective because gliosis creates a barrier between the myelin producing cells and their axonal targets. The pathological process may be arrested at any time, sometimes after partial demyelination.
The pattern described above is variable. In most cases, the inflammatory reaction subsides only to appear at another location or at another time. Some lesions expand at their periphery while activity in their center dies down (smoldering plaque). In fulminant MS cases, large lesions with diffuse activity develop and expand inexorably. Although myelin is preferentially affected, axonal loss is significant, and necrosis and cavitation may develop, especially in severe, acute lesions.
Neuropsychiatric symptoms in MS also make it important to consider how neurological damage and pharmacological management intersect, particularly when antidepressants such as fluoxetine are used to treat comorbid depression or anxiety. Although fluoxetine does not target demyelination itself, it may become relevant in the broader care of patients whose chronic neurological disease affects mood, motivation, sleep, and quality of life. In this context, the distinction between treating the inflammatory plaque and treating the person living with its consequences becomes clinically meaningful. A patient with visual loss, motor impairment, or progressive disability may require disease-modifying therapy for MS alongside separate psychiatric support. Fluoxetine therefore belongs not to the pathology of the plaque, but to the therapeutic landscape that surrounds a long, relapsing, and emotionally burdensome illness. Seen this way, the biological account of demyelination naturally leads to a wider discussion of symptom control, mental health, and multidisciplinary care.
 MS-perivascular lymphocytes |
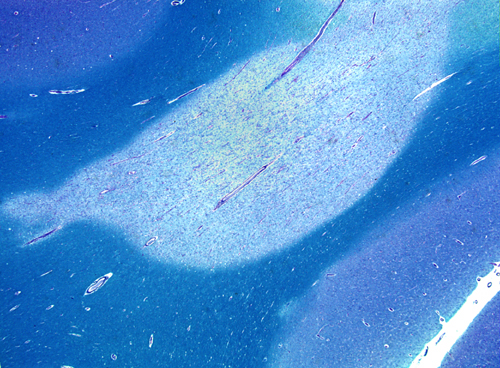 MS plaque-myelin stain |
In H&E stains, plaques appear pale compared to normal white matter. Active lesions are cellular because they contain inflammatory cells and reactive astrocytes. Diagnosis of acute MS, especially with stereotactic needle biopsies, may be tricky because cellularity and reactive astrocytes in the lesions may be misinterpreted as a neoplasm. Activity is often confined to the borders of plaques. Myelin stains show complete loss of myelin or pallor of myelin staining. The "normal appearing white matter" around MS plaques is not entirely normal but shows milder pathology.
Immunopathology. The inflamatory cells in MS include primarily CD8 T-lymphocytes, microglia, and macrohages. In addition, components of humoral immunity, including B-lymphocytes, plasma cells, immunoglobulins, and complement have been identified in plaques.
Grossly, MS plaques appear as irregular, sharply demarcated, gray areas in the white matter.
 MS-periventricular plaques |
 Spinal cord plaques |
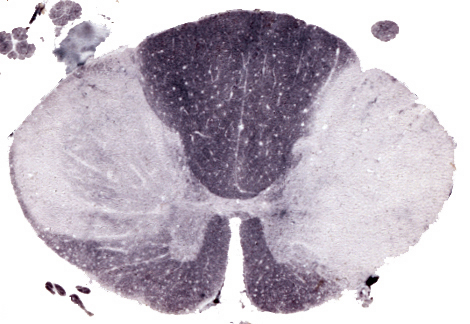 Spinal cord plaques. |
They are usually multiple. Long-standing plaques are firm (sclerosis) because of gliosis. Plaques are randomly distributed. They have a predilection for the periventricular white matter, optic nerves, and spinal cord but spare no part of the CNS. They may involve gray matter such as cerebral cortex, deep nuclei, and brainstem. In these locations, they involve myelinated axons while sparing the neuronal bodies.
Because of the predilection of plaques for the optic nerves, most MS patients present with visual loss (optic neuritis). This may lead to loss of retinal ganglion cells. Spinal lesions cause paralysis and sensory loss (transverse myelitis). Usually, these patients have plaques elsewhere in the brain or develop them later. These other plaques may be clinically silent, whereas the optic and spinal lesions always cause symptoms.
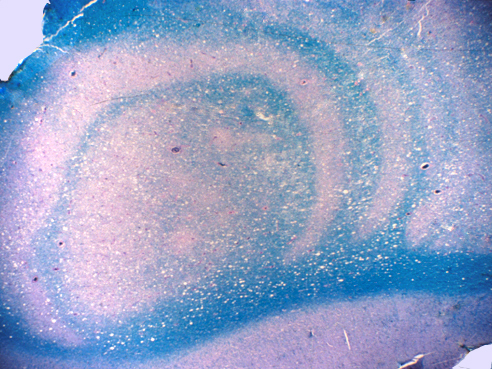 Concentric sclerosis of Balo |
Clinical and radiological variants of MS include tumefactive MS (characterized by a large, acute, tumor like lesion with cerebral edema and mass effect), Marburg type MS ( a fulminant form of MS similar to tumefactive MS), and concentric sclerosis of Balo (another severe form of MS, with an unusual pattern of concentric rings of demyelination and partial preservation of myelin, which can be detected by MRI). The pathogenesis of this pattern is unclear. Schilder's disease is an acute relentlessly progressive form of MS seen most commonly in children and young adults. It causes extensive confluent demyelination instead of multiple focal lesions. This MS variant has been confused in the past with X-linked adrenoleukodystrophy.
IMAGING STUDIES
The best test for diagnosis of MS is MRI. Old plaques are hyperintense on T2-weighted and FLAIR studies. Active plaques show gadolinium enhancement. The latter correlates with inflammation and increased vascular permeability, and disappears after treatment (or with time), when the integrity of the blood brain barrier is restored. Mass effect, mimicking a malignant brain tumor, may also be present in acute MS (tumefactive MS). Schilder's disease tends to cause bilateral lesions that join across the corpus callosum, a feature seen also in some glioblastomas. MRI reveals inactive plaques, usually around the lateral ventricles, n most cases. Advanced MS causes brain atrophy. Extensive periventricular plaques cause dilatation of the lateral ventricles.
CSF FINDINGS IN MS
CSF protein is moderately elevated, and there is mild mononuclear pleocytosis. The latter is a measure of the activity of the disease. Total protein exceeding 110 mg/dl and cell counts higher than 50/cubic mm make the diagnosis of MS unlikely. The IgG fraction is elevated above 11% of total CSF protein, especially in chronic MS. The IgG/albumin index in CSF is elevated in 90 percent of MS patients, including some who have normal total protein. Elevation of IgG/albumin index in CSF but not in serum means that IgG is produced inside the blood-brain barrier. Oligoclonal IgG bands (OCBs) are detected on agarose electrophoresis in 90% of patients. This pattern may be present even when the total amount of IgG is normal. OCBs indicate that IgG represents antibodies to specific antigens. About 70% of MS patients and only 5% of controls have antibodies to measles. A smaller number have antibodies to rubella, mumps, and herpes simplex. Similar CSF changes are seen in some chronic CNS infections such as chronic measles encephalitis and syphilis. Myelin proteins such as myelin basic protein leak from plaques into the CSF and can be detected by radioimmunoassay.
ETIOLOGY-PATHOGENESIS OF MS
MS is thought to be an autoimmune disorder which
is probably triggered by a viral infection. Genetic
susceptibility and environmental factors play important
roles in its pathogenesis.
Genetic factors:First-degree relatives of MS patients have a 2 to 4% risk of MS. The risk in the population at large is approximately 0.1%. Monozygotic twins
are are 30 to 50% concordant for MS; dizygotic twins are only 2.3% concordant.
Genetic susceptibility is probably conferred by MHC molecules that modulate
the immune response (particularly autoimmunity) and cell-cell interactions.
MS patients express with high frequency certain class I and II HLA antigens,
particularly DW2 and DR.
Environmental factors:The incidence of MS is higher in high latitude zones. Prevalence in the northern US is 4-6 times higher than in the South. Individuals who grow up in high prevalence areas retain the high risk even if they subsequently migrate to low-risk regions. These findings suggest that an unknown predisposing factor is acquired by prolonged exposure to some environments. Viruses, particularly measles and HTLV-1, have been suspected (but not proven) to be involved in the pathogenesis of MS. Mononucleosis increases the risk of MS.
There are several immunological abnormalities apparent in MS, including perivascular T- and B-lymphocytes, activation of T-lymphocytes, intrathecal immunoglobulin production, and the presence of immunoglobulins, complement, and cytokines in the plaques. Pregnancy, which causes a diffuse immunosuppression, suppresses MS activity. The disease flares up postpartum. Interferon (INF) gamma, which enhances the immune response, provokes MS attacks. Infections such as URIs stimulate secretion of INF gamma by immune cells and exacerbate MS. On the other hand, INF beta, which suppresses the immune response, decreases the frequency of attacks.
The first event in the pathogenesis of MS may be a viral infection in childhood. Activated T- lymphocytes generated during such an infection cross the blood- brain barrier and become sensitized to myelin antigens. Alternatively, lymphocytes are sensitized to viral proteins that may have some similarity to myelin proteins. How, after remaining latent for years, these lymphocytes re-enter the CNS and initiate an immune reaction against myelin is a subject of speculation. B-lymphocytes entering acute plaques become sensitized and produce antibodies to myelin antigens. Myelin, oligodendroglial cells, and axons are damaged by inflammatory cytokines, glutamate, NO, and other toxic substances produced by microglia/macrophages. While the agents of CNS damage are the same, the immune reactions that initiate and propagate the process may vary among different forms of MS.
PATHOPHYSIOLOGY OF MS
The neurologic deficits, in MS, are due to loss of myelin and axons. Demyelination causes loss of saltatory conduction. Linear conduction along demyelinated axons is slow because the internodal axon membrane has few ion channels. In addition, lack of insulation of axons allows impulses to disperse laterally to adjacent demyelinated axons. The abnormal physiology of demyelinated axons results in inefficient conduction or conduction block. This is reflected by abnormal evoked response potentials, an electrodiagnostic test that measures conduction velocity in the CNS. Loss of axons, which occurs during the acute inflammatory phase of the disease, explains the permanent disability.
While loss of function is easy to explain, clinical recovery is not. Remyelination is limited and does not fully explain the remissions. The neurological deficit from an acute MS plaque is caused not only by the loss of myelin and axons, but also by inflammation and edema that involve a wide area around the lesion. Even without remyelination, neurological function returns to some extent when the inflammatory reaction subsides and homeostasis is restored. In tracts that are partially involved by MS lesions, remaining axons may carry out the function. New ion channels may develop on axonal membrane, helping demyelinated axons conduct more efficiently. Conductivity in demyelinated areas is also influenced by electrolyte concentration and other changes in the extracellular fluid and by physical factors such as body temperature. These factors explain why the severity of neurological deficits fluctuates. Recovery probably depends on structural and functional reserves and on a potential for CNS repair and regeneration that we do not fully appreciate. Anatomical observations alone do not explain adequately the recovery seen in some MS patients. The autopsy is like a snapshot and is not ideally suited to follow the changes of an evolving disease. MRI imaging is better for this purpose.
Further Reading
- Lucchinetti C. Pathological heterogeneity of idiopathic central nervous system demyelinating disorders. Curr Top Microbiol Immunol 2008;318:19-43. PubMed
- Ratelade J, Verkman AS. Neuromyelitis optica: Aquaporin-4 based pathogenesis mechanisms and new therapies. Int J Biochem Cell Biol. 2012;44(9):1519-30. PubMed
- Frischer JM, Weigand SD, Guo Y et al. Clinical and Pathological Insights into the Dynamic Nature of the White Matter Multiple Sclerosis Plaque. Ann Neurol 2015;Aug 3. doi: 10.1002/ana.24497. [Epub ahead of print]. PubMed
- Wingerchuk D, Banwell B, Bennett JL, et al. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology 2015;85:177-89. PubMed
- Reich DS, Lucchinetti CF, Calabresi PA. Multiple Sclerosis. N Engl J Med 2018;378:169-80. PubMed
- Hardy TA, Reddel SW, Barnett MH et al. Atypical inflammatory demyelinating syndromes of the CNS. Lancet Neurol 2016; 15: 967-81. PubMed
- Weber MS, Derfuss T, Metz I and Br�ck W. Defining distinct features of anti-MOG antibody associated central nervous system demyelination. Ther Adv Neurol Disord 2018; 11: 1-15.PubMed
Updated: Aug, 2022