CENTRAL PONTINE MYELINOLYSIS (CPM)
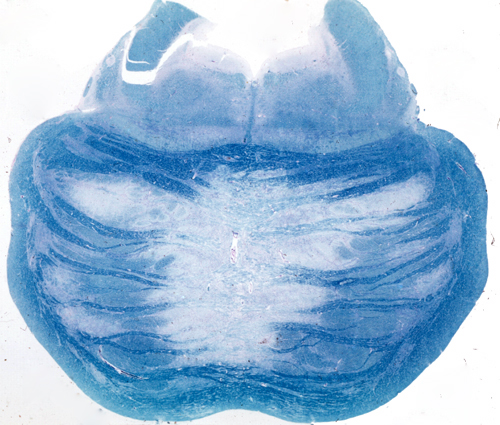 Central pontine myelinolysis |
CPM is a degeneration of a symmetrical midline patch of the basis pontis. There is loss of myelin and less severe loss of axons. Neurons of the nuclei pontis are relatively spared. No inflammation is seen. There is no selective involvement of fiber systems. In severe cases, the lesion becomes necrotic. Extension to the cerebral hemispheres and the cerebellum (extrapontine myelinolysis) is seen in some cases.
In most instances, CPM is asymptomatic. It used to be an incidental autopsy finding but now can be detected by MRI. Large lesions cause spastic bulbar paralysis, quadriplegia, stupor or coma, or the locked-in syndrome, developing in a background of severe electrolyte abnormalities.
CPM was initially reported in alcoholic patients and was thought to be a complication of alcoholism. Subsequently, it was found also in patients with burns, sepsis, cancer, liver disease, liver transplantation, malnutrition, fluid and electrolyte imbalance, and in other settings, including pediatric patients. CPM is now regarded as an osmotic demyelination syndrome. It occurs in hyponatremic patients when hyponatremia is corrected rapidly and in patients with severe hyperosmolality that was not preceded by hyponatremia. The key triggering factor is thought to be a dysosmolar state in the course of which electrolytes and organic osmolytes move out of brain cells into the extracellular space. Any further osmotic stress, at this fragile phase, may cause brain shrinkage and shearing of oligodendrocyte processes, initiating myelin break down. Experimental CPM after rapid correction of hyponatremia has been produced in dogs, rabbits, and rats.