PEROXISOMAL DISORDERS
Peroxisomes are oxidative organelles that are present in all tissues but are especially abundant in the liver and kidney. As their name indicates, they use molecular oxygen in oxidative reactions that generate hydrogen peroxide. They also contain catalase (peroxidase), which uses H2O2 to oxidize other substrates. This reaction is especially important in detoxifying ethanol, formic acid, and other toxins. Peroxisomes can be identified by light microscopy by their catalase activity using the diaminobenzidine reaction.
Peroxisomal enzymes catalyze several anabolic and catabolic reactions. The most important of these are plasmalogen synthesis and very long chain fatty acid (VLCFA) beta oxidation. Plasmalogens are the most abundant phospholipids in myelin. The products of VLCFA beta oxidation are used for biosynthesis of cholesterol, bile acids, and other compounds.
The peroxisome has a single membrane which encloses the peroxisomal matrix. The peroxisomal membrane is a lipid bilayer with embedded peroxisomal membrane proteins. All peroxisomal membrane proteins and enzymes of the peroxisomal matrix are encoded by nuclear genes, synthesized on free ribosomes, and imported into the peroxisomes. The proteins that are involved in peroxisomal biogenesis are called peroxins and are encoded by PEX genes. Fifteen human PEX genes are known.
There are two types of peroxisomal disorders: single peroxisomal enzyme deficiencies and peroxisomal biogenesis disorders (PBDs). The former are caused by mutations of genes encoding specific peroxisomal enzymes. PBDs are caused by mutations of PEX genes that are involved in the biogenesis and function of peroxisomes and are characterized by deficiencies of multiple peroxisomal enzymes and, in some cases, by absence or reduction in the number of peroxisomes. The most severe and important PBD is the Zellweger spectrum. The most frequent peroxisomal disorder is X-linked adrenoleukodystrophy (XALD). XALD is neither a single peroxisomal enzyme deficiency nor a PBD. It is due to a defect in the transport of VLCFAs across the peroxisomal membrane (see further on). The incidence of peroxisomal disorders is 1:20,000.
Most peroxisomal disorders cause severe neurological dysfunction due to CNS malformations (migration defects), myelin abnormalities, and neuronal degeneration. Non-neurological manifestations include dysmorphic features, liver dysfunction and skeletal abnormalities. The key biochemical abnormalities of peroxisomal disorders and the basis for their laboratory diagnosis are elevation of VLCFAs and decreased RBC plasmalogens.
All peroxisomal disorders except X-linked adrenoleukodystrophy are autosomal recessive. As in other genetic disorders, several mutations of each gene are seen, some of them severe and others milder. Mutations of different genes can cause similar phenotypes. The unique biogenesis of peroxisomes and multiple interactions of PEX genes explain the genetic and phenotypic complexity of peroxisomal disorders.
ZELLWEGER SPECTRUM DISORDERS (ZSD)
The Zellweger cerebrohepatorenal syndrome (ZS), neonatal adrenoleukodystrophy (NALD), and infantile Refsum disease (IRD) were initially described as separate entities. It is now clear that they represent a phenotypic spectrum, the ZSD, which is caused by mutations of 12 PEX genes resulting in abnormal peroxisomal biogenesis. The ZS is the most severe end of the spectrum and IRD the least severe.
The clinical findings of the ZS are dysmorphic features (prominent forehead, hypertelorism, epicanthal folds, flat supraorbital ridges, broad nasal bridge, large fontanelles), neurological abnormalities (hypotonia, decreased sucking, decreased tendon reflexes, seizures, nystagmus, contractures), liver disease, and calcific stippling of the patellae. Most ZS patients have failure to thrive and die by six months of age.
The general pathological changes in the ZS are cholestasis, hepatic fibrosis, and cortical renal cysts. Hepatocellular peroxisomes are absent or severely decreased. Adrenocortical cells and macrophages contain diverse lipid materials including characteristic trilamellar inclusions. The brain, in the ZS, shows neuronal migration defects (NMDs), white matter abnormalities, and lipid storage.
 ZS. Cirrhosis |
 ZS. Pachygyria |
 ZS: lipid deposits |
The most severe NMD is perisylvian pachygyria with polymicrogyria in the adjacent frontoparietal areas. The topography and cytoarchitecture of this lesion are characteristic of the ZS. The cerebellum shows microgyria and heterotopic islands of Purkinje and granular cells in the white matter. The neuronal ribbons of the dentate nuclei and inferior olives lose their elaborate folding or are discontinuous. The NMD is the main cause of the profound neurological abnormality in the ZS. The abnormality of neuronal migration may be due to impairment of cellular interactions and signaling due to incorporation of abnormal fatty acids into neuronal membranes. The white matter in the ZS is reduced in mass and shows deficient myelin. Sudanophilic lipid products accumulate throughout the brain in macrophages, neurons, and glial cells. These macrophages contain a variety of abnormal lipid products including trilamellar inclusions. These products may be gangliosides containing VLCFA and other lipids.
NALD has similar but milder dysmorphic features and neurological abnormalities to the ZS, and patients survive on average for three years and some of them into adolescence. Hepatic fibrosis and cirrhosis are common, and hepatocellular peroxisomes are normal or reduced. Adrenocortical atrophy with VLCFA storage in adrenocortical cells is identical to XALD and there are macrophages with abnormal lipid contents in many organs. The brain shows polymicrogyria, subcortical heterotopias, and cerebellar dysplasia, but no pachygyria. Lipid laden CNS histiocytes are less frequent than in the ZS. NALD patients surviving longer than three to five years develop an inflammatory demyelinative white matter disorder identical to XALD.
Infantile Refsum disease (infantile phytanic acid storage disease). IRD is characterized by psychomotor retardation, sensorineural deafness, pigmentary degeneration of the retina, anosmia, and minor dysmorphic features. Patients survive into adolescence or adulthood. The liver is enlarged and cirrhotic and hepatocytes contain lamellar inclusions similar to phytol inclusions seen in chloroplasts of plant cells. The adrenals are atrophic and the adrenal cortex contains ballooned and striated cells. MRI studies show diffuse white matter atrophy.Biochemical studies show elevation of phytanic acid, hence the term infantile phytanic acid storage (Refsum) disease. Peroxisomes are absent or reduced.
X-LINKED ADRENOLEUKODYSTROPHY
X-linked adrenoleukodystrophy (X-ALD) is caused by mutations of a gene on Xq28 that encodes a peroxisomal membrane protein, the ALD protein (ALDP). ALDP belongs to a family of ATP binding transporters and is involved in transporting VLCFA or VLCFA-CoA into peroxisomes for further processing. Without ALDP, VLCFA are not processed in peroxisomes and accumulate in glial cells including oligodendrocytes. Incorporation of VLCFA in myelin destabilizes it causing it to break down. VLCFA accumulation in the adrenal cortex causes adrenal atrophy. Adrenal insufficiency begins early in childhood. Neurological manifestations appear a few years later, usually between age five and ten. The initial abnormalities are apathy and behavioral change. Visual loss, spasticity, and ataxia follow, and patients usually die a few years after the onset of neurologic symptoms. A variant of X-ALD, adrenomyeloneuropathy (AMN), is characterized by adrenal insufficiency since childhood with progressive spastic paraparesis, peripheral neuropathy, cerebellar ataxia, and intellectual deterioration beginning in the third decade.
Neuropathologically, X-ALD is characterized by diffuse myelin loss, lipid-laden histiocytes, and perivascular lymphocytic infiltrates,especially in areas of active myelin breakdown. ALDP deficiency and VLCFA accumulation probably activate microglia and initiate an inflammatory reaction that further damages myelin.
 XALD: demyelination |
 XALD: gliosis and inflammation |
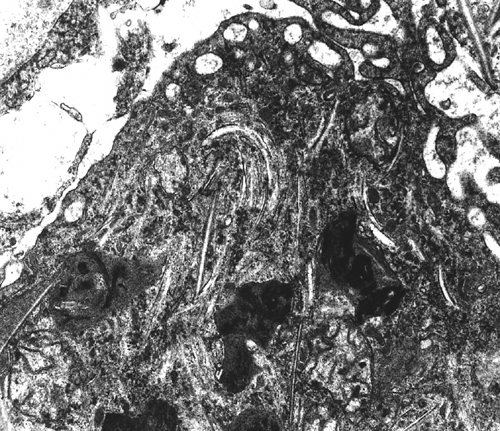 XALD: lipid deposits |
Thus, myelin loss in X-ALD is probably due to two causes, chemical imbalance and inflammation. Advanced cases show white matter atrophy and gliosis. Similar changes are seen in the advanced stages of a severe variant of multiple sclerosis, Schilder's disease, with which X-ALD has been confused in the past. Characteristic cellular inclusions (trilamellar membranes containing VLCFA cholesterol esters) are seen with the electron microscope in adrenal cortical cells, white matter histiocytes, Leydig cells, and Schwann cells.
Further Reading
- Steinberg SJ, Dodt G, Raymond GV, Braverman NE, Moser AB, Moser HW. Peroxisome biogenesis disorders. Biochim Biophys Acta. 2006;1763:1733-48. PubMed
- Moser HW, Mahmood A, Raymond GV. X-linked adrenoleukodystrophy. Nat Clin Pract Neurol. 2007;3:140-51. PubMed
Updated: May, 2017