FRONTOTEMPORAL LOBAR DEGENERATIONS
Frontotemporal dementia (FTD) is a clinical term that refers to a group of progressive neurodegenerative disorders that affect the frontal and temporal lobes causing personality change (apathy, disinhibition, loss of insight and emotional control), loss of the ability to recognize the meaning of words and objects, language dysfunction, and global cognitive decline. Three clinical FTD syndromes are recognized: behavioral variant of FTD, semantic dementia, and progressive nonfluent aphasia. Because behavior and personality change may be a presenting symptom, FTDs are often initially diagnosed as psychiatric disorders. FTDs have an earlier onset than AD and, at an early stage, do not cause the memory loss and visuo-spatial disorientation that are so characteristic of AD. There is an overlap between FTDs, amyotrophic lateral sclerosis, and atypical parkinsonian syndromes (progressive supranuclear palsy and corticobasal degeneration). The FTDs account for 10 to 20 percent of dementia, being the second or third most common cause of dementia after AD and dementia with Lewy bodies. About 40% of FTDs are familial, many of them autosomal dominant. Genes involved in autosomal dominant FTDs are MAPT, which encodes microtubule-associated protein tau, GRN (progranulin), and C9ORF72. GRN, C9ORF72, and other genes connected to the FTDs (and Parkinson's disease) regulate lysosomal function.
The term Frontotemporal Lobar Degeneration (FTLD) refers to the neuropathology of the FTDs. Pathologically, the FTLDs are characterized by atrophy of the frontal and temporal lobes (lobar atrophy) which contrasts the diffuse atrophy of AD, and by some or all of the following microscopic findings: neuronal loss and gliosis, vacuolization of the superficial cortex (spongiosis), and ballooned neurons. All FTLDs show also abnormal protein inclusions in neurons and glial cells. Based on the chemical nature of these inclusions, three groups of FTLDs are recognized:
- FTLD-tau (MAPT mutations): neuronal (Pick bodies) and glial cytoplasmic inclusions, NFT-like structures.
- FTLD-TDP (GRN mutations and C9ORF72 expansions): tau-negative, alpha-synuclein-negative cytoplasmic and intranuclear inclusions of ubiquitinated TAR DNA-Binding protein 43 (TDP-43) in neurons and glial cells. C9ORF72 localizes in lysosomes and regulates autophagic function.
- FTLD-FUS (mutations of FUS): neuronal cytoplasmic and intranuclear inclusions containing fused sarcoma protein (FUS).
Though degeneration in FTLDs is primarily cortical, there are pathological changes in the substantia nigra and other subcortical structures. The pathological diagnosis of FTLDs requires extensive tissue sampling and immunohistochemistry targeting tau, TDP-43, and ubiquitin.
Our concept of FTDs and FTLDs is still evolving but significant progress has been made recently in sorting out these entities and other non AD dementias and getting insights into their pathogenesis and genetics.
FTLD-TAU (TAUOPATHIES)
Tau (the Greek letter) is a microtubule associated protein, encoded by the gene MAPT gene on 17q21. Normally, tau is phosphorylated and is present mainly in axons where it binds and stabilizes microtubules. In FTLDs, abnormal deposits of hyperphosphorylated tau in the form of paired helical or straight filaments are deposited in neurons and glial cells. The FTLD-TAU group includes:
Pick's Disease FTD with Parkinsonism Linked to Chromosome 17 (FTDP-17), a MAPT mutation Corticobasal Degeneration Progressive supranuclear palsy |
|---|
AD is also in part a tauopathy. In AD, the tau deposits (NFTs and neuropil threads) are present in the neuronal body and dendrites. In the FTLDs, both, neurons and glial cells are affected.
PICK’S DISEASE
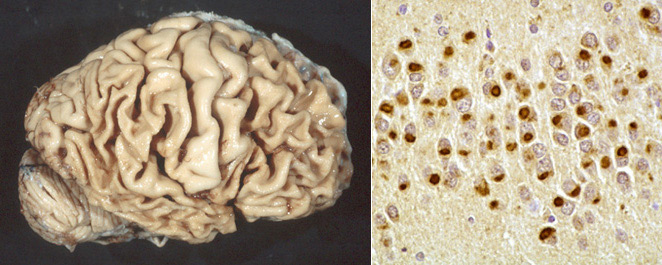 Pick's disease |
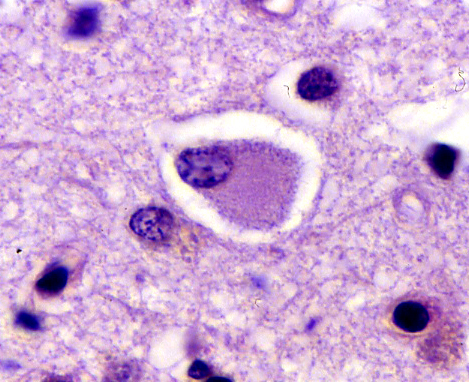 Ballooned neuron (Pick cell) |
Described almost 2 decades before AD, Pick's disease has long been the prototype of FTLDs. It presents between 45-65 years with confusion and FTLD symptoms and has a progressive course lasting 2-5 years, sometimes more. In advanced stages it cannot be distinguished clinically from AD. The brain shows atrophy of frontal and temporal lobes, Pick bodies (tau-positive spherical cytoplasmic neuronal inclusions, composed of straight filaments), and Pick cells (ballooned neurons with dissolution of chromatin). Older patients may also have AD pathology.
TDP-43 PROTEINOPATHIES
Transactive response RNA-binding protein 43kDa (TDP-43), encoded by the TARDBP gene,
is a highly conserved nuclear protein, which is expressed in many tissues.
Its functions have not been completely defined. Mutations of TARDBP cause familial FTD and ALS.
In these diseases, abnormally folded, hyperphosphorylated TDP-43, conjugated with ubiquitin, is deposited
in neurons in the form of intranuclear, cytoplasmic, and neuritic inclusions. The core clinical and pathological
phenotype of the TDP-43-associated FTD is similar to that of other FTDs. Clinically, a significant proportion
of ALS patients present with FTD symptoms or develop cognitive impairment later. Pathologically,
TDP-43 inclusions are present in degenerating motor neurons in ALS and are also seen in the cortex and other
locations. TDP-43 deposits are also seen in Parkinson’s disease, Guam Parkinson dementia complex,
progressive muscular atrophy, primary lateral sclerosis, some cases of AD, inclusion body myopathy
and traumatic brain injury. TDP-43 binds RNA and is involved in its processing. TDP-43 proteinopathies may be
caused by loss of function mutations affecting protein synthesis or gain of function leading to abnormal aggregation.
Further Reading
- Kwong LK, Uryu K, Trojanowski JQ, Lee VM-Y. TDR-43 Proteinopathies: Neurodegenerative Protein Misfolding Diseases without Amyloidosis. Neurosignals 2008:16;41-51. PubMed
- Josephs KA. Frontotemporal Dementia and Related Disorders: Deciphering the Enigma. Ann Neurol 2008;64:4-14. PubMed
- Seltman RE, Matthews BR. Frontotemporal Lobar Degeneration. Epidemiology, Pathology, Diagnosis and Management. CNS Drugs 2012; 26:841-870. PubMed
- Bigio EH. Making the Diagnosis of Frontotemporal Lobar Degeneration. Arch Pathol Lab Med 2013;137:314-35. PubMed
- Mann D, Snowden JS. Frontotemporal lobar degeneration: Pathogenesis, pathology and pathways to phenotype. Brain Pathology 2017; 27:723–736. PubMed
Updated: May, 2020