INFLAMMATORY MYOPATHIES
Inflammatory myopathies (IMs) are acquired, treatable autoimmune diseases, characterized clinically by weakness and soreness of muscles and elevated CK and pathologically by myonecrosis and mononuclear inflammatory infiltrates. A large number of autoantibodies are associated with IM and some cases overlap with connective tissue disorders and involve other organs besides skeletal muscle. Five IMs are now recognized based on clinical symptoms and autoantibody profiles: Overlap myositis (OM), dermatomyositis (DM), autoimmune necrotizing myositis (NM), polymyositis (PM) and inclusion body myositis (IBM). Each of these are probably not distinct entities but rather syndromes with variable phenotypes and autoantibody patterns. DM affects children and adults. All other IMs are adult disorders. IBM is clinically and pathologically distinct (see below). DM also has distinct clinicopatholological features but some cases overlap with OM and PM. The diagnosis of IM is based on the clinical features (mode of onset and progression, pattern of weakness, extramuscular manifestations, CK levels), analysis of autoantibodies, and muscle biopsy. An important and useful diagnostic feature of IM is the expression of HLA-1 (MHC-1) by myofibers. Normally, MHC-1 is expressed in muscle capillaries but not in myofibers. In IM, MHC-1 is expressed in the sarcolemma and sarcoplasm of all myofibers, even if there is no inflammation. MHC-1 expression is also seen in some muscular dystrophies. More muscle biopsies are done for diagnosis of IM than for any other category of muscle disease.
 Mixed CTD |
 Autoimmune necrotizing myositis |
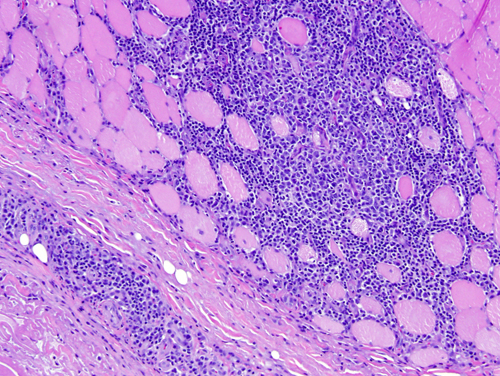
Overlap myositis, a.k.a. mixed connective tissue disease (CTD), undifferentiated CTD, diffuse CTD, and systemic rheumatic disease refers to IM combined with features of systemic lupus erythematosus, scleroderma and other CTDs. The muscle biopsy in such cases shows primarily interstitial inflammation that resembles DM. OM patients may also have cardiac involvement leading to arrhythmia and heart failure, arthralgia, Raynaud's phenomenon, interstitial pneumonitis, and renal involvement. Some of these extra-muscular manifestations are associated with circulating anti-Jo-1 (anti tRNA synthetase) and other autoantibodies.
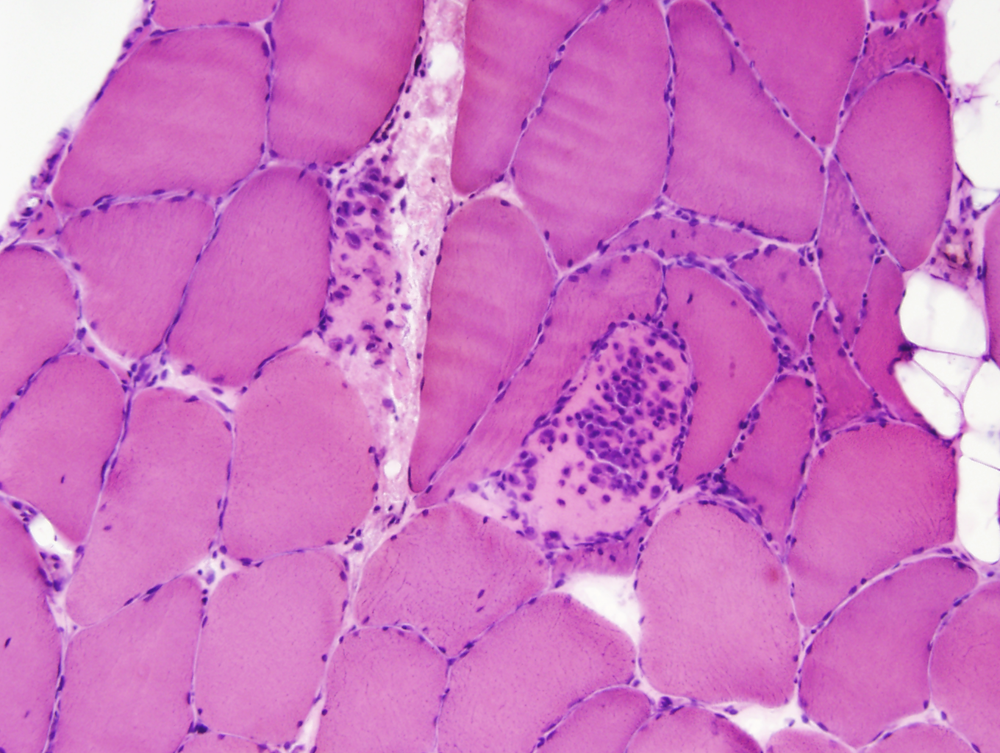
Immune-mediated necrotizing myopathy (IMNM) is a recently recognized IM associated with anti-signal recognition particle (SRP) and anti-hydroxy-3-methylglutaryl-CoA reductase (HMGCR) autoantibodies. It occurs in children and adults, has an acute or subacute onset and causes proximal arm and leg weakness. It is more severe than DM and PM and is associated with marked CK elevation. Extra-muscular manifestations such as cardiac involvement and interstitial lung disease are infrequent. The muscle biopsy in IMNM shows myofiber necrosis and regeneration with minimal or no inflammation. Patients with weakness, CK elevation and myonecrosis on muscle biopsy are presumed to have antibody-negative IMNM. Slowly progressive, antibody-negative IMNM myopathy in children may mimic limb-girdle progressive muscular dystrophy.
Dermatomyositis affects children and adults and is the most common inflammatory myopathy in children. It presents subacutely with muscle pain, weakness, and stiffness, and causes also a purple (heliotrope) discolorarion of the upper eyelids, edema around the eyes and mouth, facial erythema, and erythematous scaly papules over the knuckles and elbows (Gottron’s sign). Contractures, subcutaneous calcification, intestinal ulceration, and other extramuscular manifestations are frequent in children. Adult DM is frequently associated with CTD and cancer.
 Dermatomyositis |
 Necrotic capillary |
 Tubuloreticular inclusion |
The pathology of DM includes inflammation, vasculitis, and perifascicular atrophy. The inflammatory cells are predominantly B-cells (with smaller numbers of CD4-positive T-cells) and are found around blood vessels, in the septa between muscle fascicles, and in fibroadipose tissue around muscle. The key pathological change of DM is a vasculitis, which involves endomysial and perimysial capillaries and arterioles. This vasculitis begins with endothelial swelling and is followed by endothelial necrosis and capillary loss. Tubuloreticular cytoplasmic inclusions (TRIs) are often seen in endothelial cells. TRIs also occur in lupus and other collagen vascular diseases but are absent in PM and IBM. The vasculitis is thought to be caused by circulating anti-endothelial antibodies. Interaction of these antibodies with vascular antigens activates complement, leading to formation of the membranolytic attack complex (MAC), which destroys endothelial cells.
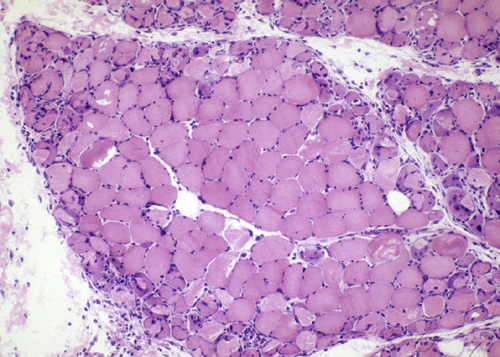 Perifascicular atrophy |
 Perifascicular atrophy |
A distinctive feature of DM is atrophy and degeneration of myofibers at the periphery of fascicles (perifascicular atrophy-PFA), which occurs even in absence of inflammation. It has been proposed that PFA is caused by ischemia from loss of the endomysial capillary bed. This affects more severely the distal portion of the vascular field, which is the periphery of each fascicle. In support of this hypothesis, ischemic infarction of muscle is seen in some cases of DM. However, PFA is not seen in diabetic and other angiopathies. Recent evidence indicates that the CD4-positive cells in DM are plasmacytoid dendritic cells. These cells secrete type 1 interferons, which induce genes and molecular cascades that cause muscle injury. A key interferon-induced pathology is deficiency of titin, an intramuscular protein that is a scaffold for contractile filaments. It is now thought that diffusion of interferons in the perifascicular areas causes the PFA.
Dermatomyositis and polymyositis (and less frequently inclusion body myositis) are associated with scleroderma, mixed connective tissue disease, and cancer. The association with cancer is stronger with dermatomyositis. Patients with polymyositis and dermatomyositis may also have cardiac involvement leading to arrhythmia and heart failure, arthralgia, Raynaud's phenomenon, interstitial pneumonitis, and renal involvement. Some of these extra-muscular manifestations are associated with circulating antibodies to anti-Jo-1 (an anti tRNA synthetase) autoantibodies.
 Polymyositis |
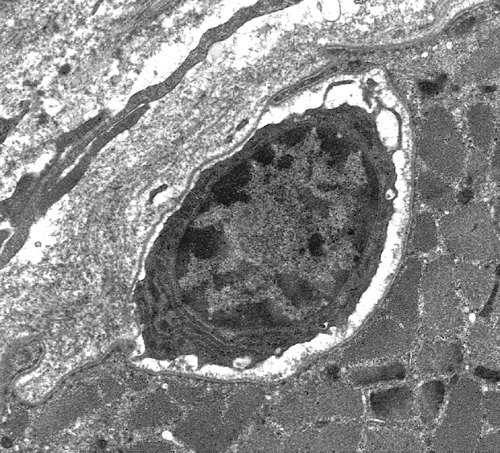 Lymphocyte in myofiber |
Polymyositisis now thought to be rare and is diagnosed by exclusion of other IMs. It affects predominantly adults who present with proximal weakness (without a rash), developing over a period of weeks or months. The muscle biopsy shows endomysial mononuclear cells and myonecrosis. PM is a cell-mediated autoimmune disorder in which cytotoxic (CD8-positive) lymphocytes and macrophages invade and destroy myofibers. The inflammatory cells are in the endomysium (between and around individual myofibers.) Inflammation may be focal and the MRI is useful in identifying affected areas for biopsy. Mild inflammation may also be present in some muscular dyhstrophies.
 Inclusion body myositis |
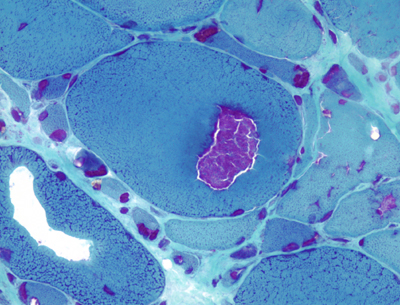 Inclusion body myositis |
 Inclusion body myositis |
Inclusion body myositis is the most common muscle disease in old people. It has an insidious onset and causes slowly progressive proximal and distal weakness with mild CK elevation. The pathology of IBM is highly characteristic and combines inflammation and myofiber degeneration. The inflammation is similar to PM with cytotoxic (CD8-positive) lymphocytes invading and destroying myofibers. The degenerative changes consist of accumulation of myelinoid bodies and amyloid deposition. Affected myofibers have vacuoles or cracks, which contain basophilic granules. These are best seen in cryostat sections stained with modified Gomori trichrome. The granules consist of myelinoid membranous bodies. In or near the vacuoles there are small chunks of amyloid that can be detected with Congo Red stains. With electron microscopy, the amyloid deposits contain paired helical filaments similar to the neurofibrillary tangles of Alzheimer's disease and straight filaments. The filamentous inclusions of IBM contain beta amyloid, hyperphosphorylated tau protein (best detected with antibodies to SMI-31), apolipoprotein E, presenillin 1, prion protein, α-synuclein, and other proteins. Some of these are the same proteins that accumulate in the brain in neurodegenerative diseases such as Alzheimer's disease and Parkinson’s disease. Ragged red fibers and Cytochrome C-negative fibers are present in IBM with higher frequency than similar age individuals without IBM. The pathogenesis of IBM is not known but probably involves abnormal protein processing associated with ageing of myofibers, and the deposition of toxic protein polymers that damage myofibers and trigger inflammation. IBM is refractory to immunosuppressive therapy that is used in other inflammatory myopathies. There are hereditary myopathies with similar myofiber changes but without inflammation. The pathogenesis of IBM is not known but probably involves abnormal protein processing associated with ageing of myofibers, and the deposition of toxic protein polymers that damage myofibers and trigger inflammation. IBM is refractory to immunosuppressive therapy that is used in other inflammatory myopathies. There are hereditary myopathies with similar myofiber changes but without inflammation.
PM and DM are treated with corticosteroids. Azathioprine, methotrexate, and cyclosporin are used in severe cases. IBM is refractory to corticosteroids and immunosuppressive agents. The inflammation of PM, DM, and collagen vascular disease subsides rapidly with corticosteroids, so the biopsy should be done before treatment is started.
MYASTHENIA GRAVIS
Myasthenia gravis is an autoimmune muscle disease characterized by progressively increasing weakness with exertion and recovery of strength with rest or following administration of anticholinesterase drugs such as neostigmine. Weakness affects most severely muscles that are innervated by brain stem nuclei, such as extraocular and facial muscles, and causes drooping of the eyelids, diplopia, and inability to chew. Death is due to respiratory compromise. Pathologically, the muscle is either normal or shows myofiber atrophy and aggregates of lymphocytes in the endomysium. In severe cases, there may be myonecrosis. Electron microscopy shows abnormal motor end plates. Axon terminals are normal and the number of synaptic vesicles is adequate, but there is loss of post-synaptic membrane such that the post-synaptic region is simplified, showing a few wide folds without branching. The primary synaptic cleft is widened. Ten percent of patients with myasthenia gravis, especially older males, have thymomas, and most other patients have follicular hyperplasia of the thymus.
Myasthenia gravis is caused by antibodies to the acetylcholine receptor (AChR) protein. Circulating IgG antibodies bind with the AChR and prevent acetylcholine from reacting with it. These antibodies also cause degradation of AChR and lysis of post-synaptic membranes. Improvement of strength following administration of edrophonium (Tensilon test) or neostigmine are diagnostic. Treatment consists of anticholinesterase drugs, corticosteroids and thymectomy.
Further Reading
- Dalakas M. Polymyositis and dermatomyositis. Lancet 2003; 362:971-82.PubMed
- Mammen AL. Dermatomyositis and polymyositis. Clinical presentation, autoantibodies, and pathogenesis. Ann NY Acad Sci 2010; 1184:134-153. PubMed
- Salajegheh M, Kong SW, Pinkus JL, et al. Interferon-Stimulated Gene 15 (ISG15) Conjugates Proteins in Dermatomyositis Muscle with Perifascicular atrophy. Ann Neurol 2010;67:53-63. PubMed
- Askanas V, Engel WK, Nogalska A. Pathogenic considerations in sporadic inclusion-body myositis, a degenerative muscle disease associated with aging and abnormalities of myoproteostasis. J Neuropathol Exp Neurol. 2012 Aug;71(8):680-93.PubMed
- Schmidt J. Current Classification and Management of Inflammatory Myopathies. Journal of Neuromuscular Diseases;2018:109–129PubMed
Updated: March, 2024