MALFORMATIONS OF CORTICAL DEVELOPMENT AND NEURONAL MIGRATION DEFECTS
NORMAL CORTICAL DEVELOPMENT
The neurons and glial cells that form the cerebral cortex are generated around the ventricles of the brain and migrate to the cortex. Proliferating multipotential precursor cells form a thick layer around the ventricles, the proliferative neuroepithelium. The first wave of migration results in formation of a provisional cortex, the pre-plate. This is replaced by the permanent cortical plate. Glutamatergic neurons migrate to the cortex along a scaffold of cellular processes, the radial glia. The nuclei of the radial glial cells are located in the walls of the ventricles and their processes stretch vertically to the pial surface. In addition to providing guidance, the radial glial cells are also neuronal and glial precursors. Migrating neurons are guided by adhesion molecules that are present on their membranes and on radial glial fibers, and by chemical signals, some of which are produced by the pre-plate. The neurons that form the permanent cortical plate migrate in an inside out, outside last pattern. GABAergic (inhibitory) neurons are generated in the primordia of the basal ganglia and migrate to the cortex in a tangential direction. The proliferative neuroepithelium produces more neurons than are necessary to populate the cerebral cortex. Neurons that do not make working synapses die. Other neurons are eliminated by genetically programmed apoptosis.
Neurogenesis in fish, amphibians, birds, and rodents continues after birth. Until recently, it was thought that neurogenesis and migration in primates is completed by mid-gestation except for the hippocampus and the granular layer of the cerebellum, where it continues during early post-natal life. Recent research shows that neurogenesis also occurs in the adult brain. New neurons are generated in the periventricular area and migrate to the neocortex. The significance of this observation in terms of neuronal plasticity and CNS repair is not known.
Cortical development entails the generation of stem cells, their differentiation into neurons and glia, migration to the cortex, and organization into functional layers. These processes overlap are orchestrated by multiple genetic pathways. Impairment of any of these processes can cause a variety of abnormalities the major types of which are listed below. The term Neuronal Migration Defects (NMDs) is used to define some of these abnormalities but the broader term Malformations of Cortical Development (MCDs) is more inclusive.
MALFORMATIONS OF CORTICAL DEVELOPMENT
| ABNORMAL NEURONAL-GLIAL PROLIFERATION OR APOPTOSIS Microcephaly Megalencephaly |
| ABNORMAL NEURONAL MIGRATION Periventricular nodular heterotopia Lissencephaly/subcortical band heterotopia Cobblestone cortex/congenital muscular dystrophy |
| ABNORMAL CORTICAL ORGANIZATION Polymicrogyria Focal Cortical Dysplasia |
Five outcomes are possible in the MCDs:
1. Fewer or more than normal neurons are produced (microcephaly, megalencephaly).
2. Neurons
do not migrate at all from the ventricles (periventricular
heterotopia) or migrate half way (subcortical band
heterotopia).
3.
Some neurons
reach the cortex but large numbers do not. No normal
cortical layers are formed (lissencephaly, pachygyria,
cobblestone cortex).
4.
Neurons over-shoot the
cortex and end up in the subarachnoid space (marginal-leptomeningeal
glioneuronal heterotopia, cobblestone cortex).
5. The late stage of migration and cortical organization
is disrupted (polymicrogyria).
Any of these outcomes, alone or in combination, may occur in the MCDs. The process of neuronal migration and cortical organization is tied to the process of cortical folding. Abnormal migration causes an abnormal gyral pattern. The nomenclature of the MCDs reflects the naked eye appearance of the brain. In addition to abnormal folding, the cortex is usually thick and disorganized. Severe MCDs such as lissencephaly and polymicrogyria are associated with psychomotor retardation and intractable seizures.
Most MCDs have a genetic basis, and numerous genes, chromosomal loci, and genetic syndromes associated with MCDs are known. Genotypes and phenotypes overlap. The same gene mutation can cause different phenotypes because a) mutations affect protein function differently and b) the effect of a given mutation may be moderated by somatic mosaicism or, in the case of X-linked genes, by skewed X inactivation (see below). Also, mutations of different genes cause a similar phenotype, probably by influencing common pathways involved in neuronal migration and cortical development. In the past, the classification of MCDs was based on the phenotype. Now, gene defects are integrated into the classification schemes and one day may be the sole basis of classification.
The best known MCD in animals is the reeler, a naturally occurring mouse mutant in which the normal inside-out pattern of migration does not occur and each layer of migrating neurons is deposited below the previous one. As a result, the cortical layers are inverted. This MCD is caused by mutations of reelin, a protein secreted by the Cajal-Retzius cells of the preplate. Reelin is an important guidance signal for migrating neurons. The neuropathology and genetics of the major MCDs in humans are described below.
PATHOLOGY AND GENETICS OF MCDs
Microcephaly
Microcephaly, defined as a head circumference 2-3 SD below the mean, is a condition most frequently associated with subnormal intelligence and can be acquired or genetic. Acquired (secondary) microcephaly is caused by prenatal or perinatal brain injury due to hypoxic/ischemic insults, congenital infections, and other causes. Most cases of microcephaly, however, have a genetic cause. Microcephaly is a frequent component of MCDs and other brain malformations and inherited metabolic disorders. The pathology in these cases reflects the underlying condition. Several chromosomal abnormalities and genetic syndromes are associated with microcephaly. These cases have additional pathological abnormalities involving the brain and other organs. Other cases of genetic microcephaly have just small brains without other pathology. Most of these cases are caused by mutations of genes that are important in the mitotic apparatus (centrosomes, mitotic spindle), indicating that the key abnormality is defective neurogenesis. Cilia are important for cell division, and some of these mutations affect cilia function. Another set of genetic microcephaly cases are caused by gene defects affecting DNA repair.
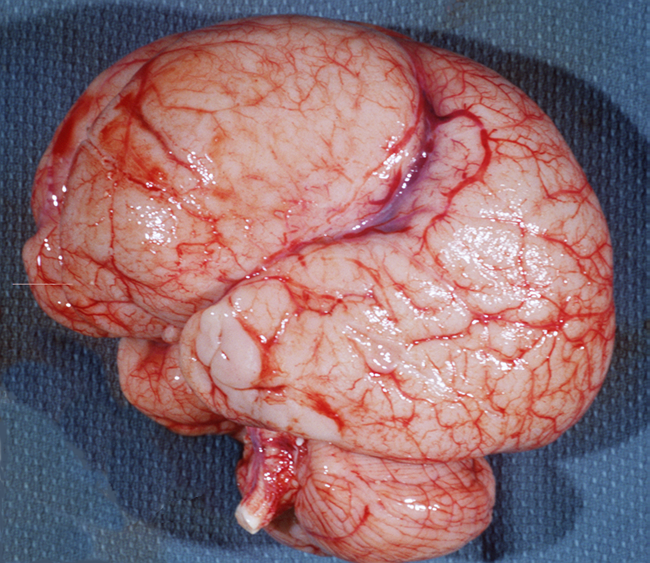
Megalencephaly-Hemimegalencephaly
 Thanatophoric dysplasia |
 Megalencephaly |
 Hemimegalencephaly |
Megalencephaly (brain weight exceeding 1800 gm or 3 SD above the mean) is a heterogeneous entity. Megalenephaly due to white matter edema or accumulation of storage material occurs in some leukodystrophies (Canavan’s disease, Alexander’s disease) and lysosomal storage diseases. Some patients have just large but otherwise normal brains. In other cases, large brain size is accompanied by polymicrogyria and other types of MCDs. A distinct form of MCD characterized by megalencephaly, deep temporal sulci, agenesis of the hippocampus, and other dysplastic changes affecting the temporal lobes occurs in thanatophoric dysplasia. In hemimegalencephaly, one hemisphere is normal and the other hemisphere (or parts of it) is enlarged and shows severe cortical abnormalities, including pachygyria, polymicrogyria, and changes similar to type 2 focal cortical dysplasia (see below). Patients with megalencephaly without cortical dysplasia may be asymptomatic or have developmental delay and mild cognitive abnormalities. Patients with cortical dysplasia have severe psychomotor delay and seizures. Dysregulation of the MAPK and mTOR pathways account for most cases of megalencephaly and hemimegalencephaly. Megalencephaly without cortical dysplasia occurs as an autosomal dominant trait and in several syndromes, including NF1, Sotos, Cowden, and other. Megalencephaly with cortical dysplasia occurs in several syndromes and gene mutations. Hemimegalencephaly is also a component of several syndromes, including Proteus, TSC, NF1, and the Clippel-Trenaunay syndrome, which is characterized by hypertrophy and vascular abnormalities affecting one side of the body.
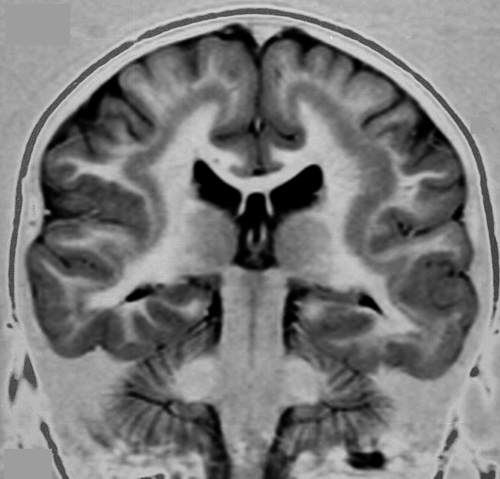
Periventricular Nodular Heterotopia (PNH)
 Periventricular heterotopia |
 Periventricular heterotopia |
PNH is characterized by unorganized islands of neurons under the ependyma of the lateral ventricles. These neurons presumably failed to migrate, and differentiated in their original positions. Unilateral PNH occurs frequently with other MCDs and brain malformations, and may be an incidental finding in patients undergoing MRI testing for unrelated reasons.
Bilateral PNH (BPNH) is caused, most commonly, by mutations of the FLNA gene, located on Xq28. The product of this gene, filamin-A, is an actin-binding protein that cross-links actin filaments and is important for the integrity of the cytoskeleton and cellular movement. BPNH is X-linked dominant, and is lethal in males. Because of random inactivation of the X chromosome in each cell, females have two populations of neurons: one normal, which migrates to the cortex, and one mutant, which does not. Females with BPNH have normal intelligence but may have seizures.
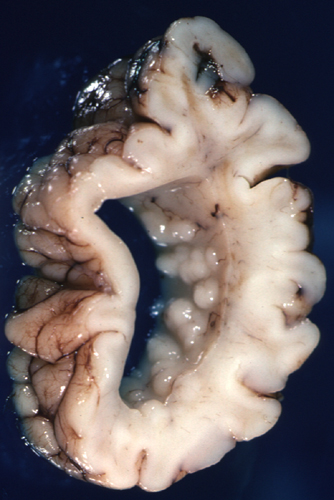
Lissencephaly-Agyria (type 1 lissencephaly)
 Lissencephaly |
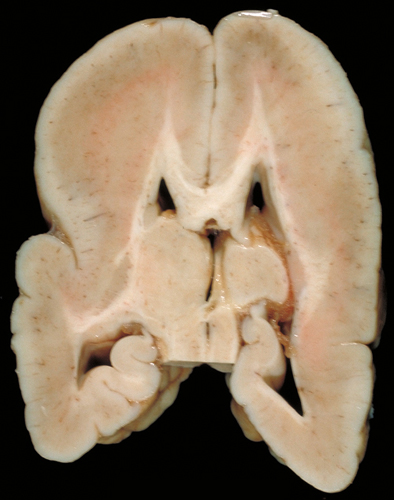 Lissencephaly |
 Lissencephaly |
 Lissencephaly |
 Pachygyria |
Lissencephaly means smooth brain. Cortical sulci in lissencephaly are absent except, usually, for the Sylvian fissure. The cortex is thick and consists of 4 layers. Layer I is the molecular layer. Layer II is a layer of pyramidal cells. Layer III is thin band of white matter with sparse neurons. Layer IV which is the thickest and most cellular, is comprised of neurons that migrated a certain distance from the ventricles but failed to reach their normal destinations. There is a small amount of myelinated white matter between the abnormal cortex and the ventricles. The cortical cytoarchitecture may vary from four layers to two, and the gyral pattern may vary from total absence of gyri to pachygyria. Patients with lissencephaly have severe psychomotor retardation and intractable seizures. Pachygyria is a milder variant of lissencephaly, characterized by broad gyri and a thick cortex with an abnormal cytoarchitecture.
Type 1 lissencephaly is caused by:
LIS1 mutations. Complete loss of LIS1 (on 17p13) is fatal. Deletion of one copy causes lissencephaly. The LIS1 protein forms complexes with other proteins that are crucial for cell division, migration, and intracellular transport. It is involved in platelet function. It localizes also in microtubules and may play a role in their regulation. Deletions of LIS1 and contiguous genes causes the Miller-Dieker Syndrome, which combines lissencephaly with dysmorphic facial features, visceral abnormalities, and polydactyly. Lissencephaly, in LIS1 mutations, is more severe in the occipital lobes. Mosaic LIS1 mutations cause subcortical band heterotopia (see below).
 Subcortical band heterotopia |
Tubulinopathies and related MCDs. Mutations of 9 tubulin and related genes cause a spectrum of NMDs ranging from severe lissencephaly to polymicrogyria. These genes function during many phases of brain development including neurogenesis, neuronal migration, and differentiation. The most severe forms of these MCDs are associated with cerebellar hypoplasia and agenesis of the corpus callosum. Patients with these MCDs have severe psychomotor retardation and intractable seizures.
X-linked lisssencephaly with ambiguous genitalia (XLAG). This form of lissencephaly, which is associated also with agenesis of the corpus callosum, is caused by mutations of ARX, which normally regulates other genes involved in neurogenesis, neuronal migration, and axonal guidance. While LIS1 and XLIS affect radial migration, ARX mutations involve tangential migration, and produce a different cortical pattern.
RELN and VLDLR mutations. The RELN gene, on 7q22, encodes a protein that binds with the very low-density lipoprotein receptor and other molecules that are involved in guidance of migrating neurons. In humans, RELN and VLDLR mutations cause pachygyria with extreme cerebellar hypoplasia. RELN mutations cause the reeler mutant mouse, characterized by cerebellar hypoplasia and cerebral cortical malformation.
Cobblestone cortex (type 2 lissencephaly)
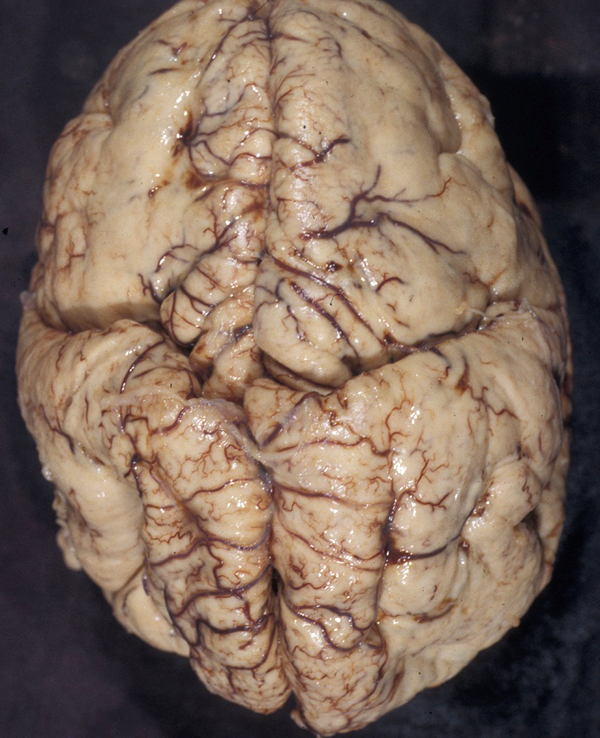 Cobblestone cortex |
 Cobblestone cortex |
 Cobblestone cortex |
This MCD is distinct from type 1 (classic) lissencephaly. The cortical surface displays irregular grooves imparting a cobblestone pattern. No cortical layers are present. Instead, there is an irregular scramble of neurons and molecular layer. The pia is interrupted, and neuroglial tissue spills into and obliterates the subarachnoid space. The lateral ventricles are dilated. Patients with cobblestone cortex have severe psychomotor retardation, seizures, visual loss, and congenital muscular dystrophy.
Cobblestone lissencephaly occurs in several genetic diseases, the most common of which are the Walker-Warburg Syndrome, Fukuyama congenital muscular dystrophy, and muscle-eye-brain disease. These conditions have a wide phenotypic spectrum and are caused by mutations of several genes that are involved in O-glycosylation of α-dystroglycan. Defective glycosylation of matrix proteins in the brain presumably impairs the interaction of migrating neurons with matrix elements, causing MCDs. In muscle, defective glycosylation of α-dystroglycan causes congenital muscular dystrophy.
Polymicrogyria
Generalized PMG |
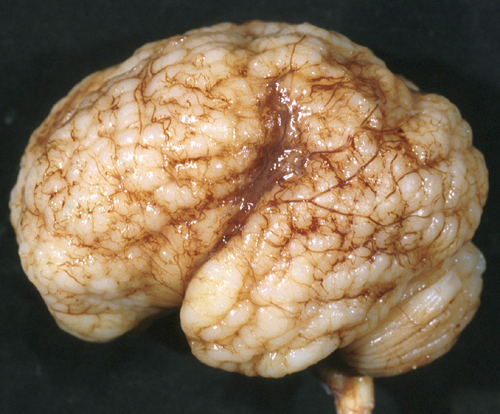 Generalized PMG |
 PMG- MCA territory |
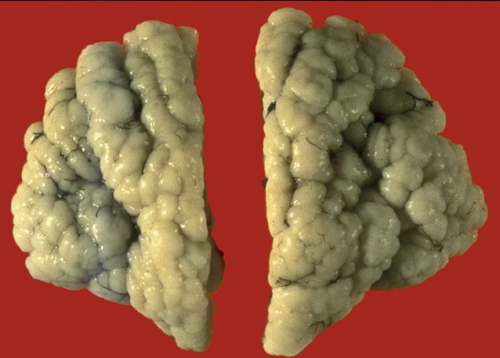 Polymicrogyria |
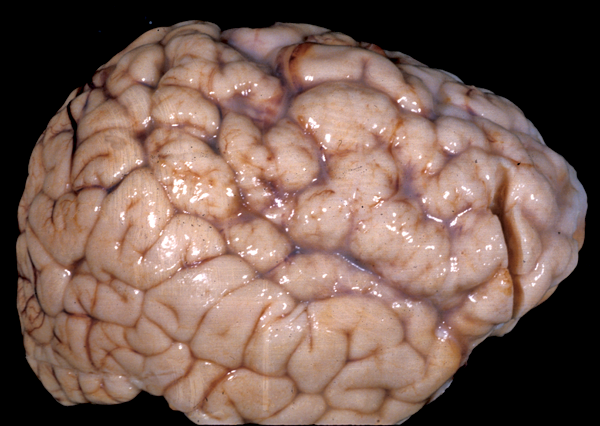 Perisylvian polymicrogyria |
In polymicrogyria (PMG), the surface of the cerebral hemispheres shows multiple small bumps -it has been likened to Morocco leather- suggesting an excessive number of gyri. The number of cortical layers varies from 2 to 4 (most commonly 4). Different layer patterns may be present in the same brain.
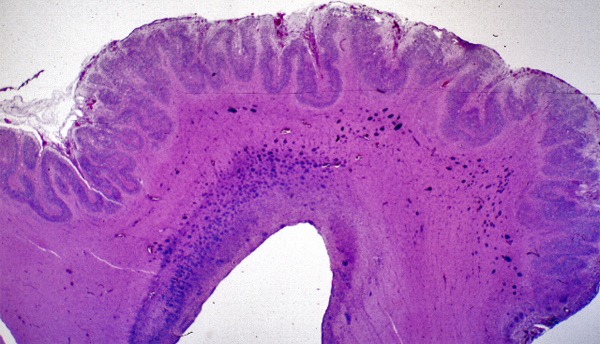 Polymicrogyria |
The cortical layers are irregularly over-folded (festooned) and the molecular layer is fused, eliminating sulci and trapping leptomeningeal vessels. PMG can be diffuse or focal, bilateral or unilateral, symmetric or asymmetric. About two thirds of PMG are perisylvian. Other topographies are less common. Some cases of PMG have also periventricular nodular heterotopias and other brain malformations, such as abnormal Sylvian fissures, agenesis of the corpus callosum, and cerebellar hypoplasia. Patients with PMG may have hemiparesis or quadriparesis, seizures, severe psychomotor retardation, and other neurological abnormalities, depending on the type, localization, and extent of the pathology.
The etiology and pathogenesis of PMG are diverse. Some forms of PMG are acquired and are caused by prenatal hypoxic-ischemic insults. In such cases, layer five is damaged and layers superficial to it overfold and fuse. The damage probably occurs either after neuronal migration is completed or following the migration of layer six neurons. In the latter case, layer four, three, and two neurons pass through the damaged layer five and are arranged in an abnormal fashion superficial to it. PMG is frequently seen in vascular territories, especially the MCA, or watershed areas, and in the borders of porencephalic cysts and schizencephaly. Some patients have schizencephaly in one hemisphere and PMG in the same vascular territory in the other hemisphere. PMG is also frequently seen in prenatal CMV and less frequently other infections. CMV has a predilection for the walls of the lateral ventricles and, if it occurs early in gestation, destroys stem cells and radial glial cells located in this zone. Destruction of these critical elements can impair neurogenesis and neuronal migration.
Other forms of PMG have a genetic basis and are associated with chromosomal deletions and mutations of WDR62, SRPX2, PAX6, TBR2, TUBB2B, and several other genes. Several genetic syndromes, including the DiGeorge syndrome, the Aicardi syndrome, and others are associated with PMG. Syndromes of bilateral symmetrical PMG have been described. The best known are bilateral perisylvian and bilateral frontoparietal PMG. Bilateral perisylvian PMG may be familial or sporadic and is due to mutations of SRPX2 and other loci on 22q11 and Xq28. It causes pseudobulbar palsy, seizures, mental retardation, motor deficits, and other symptoms. Mild cases may have dysphasia and dyslexia and few other manifestations. Bilateral frontoparietal PMG is due to mutations of GPR56 and causes developmental delay, seizures, and cerebellar and brainstem dysfunction. The pathology resembles cobblestone lissencephaly. The lateral ventricles are large and there is periventricular whie matter signal abnormality. The brainstem is small and the cerebellum is dysplastic.
MCDs in metabolic disorders
A significant number of metabolic disorders are associated with MCDs. These include peroxisomal disorders (Zellweger syndrome, neonatal adrenoleukodystrophy) associated with pachygyria-polymicrogyria, mitochondrial disorders, disorders of glycosylation associated with cobblestone cortex, and several amino acid disorders and organic acidurias (non-ketotic hyperglycinemia, histidinemia, glutaric aciduria type 2, fumaric aciduria, maple syrup urine disease, and other).
Focal cortical dysplasia
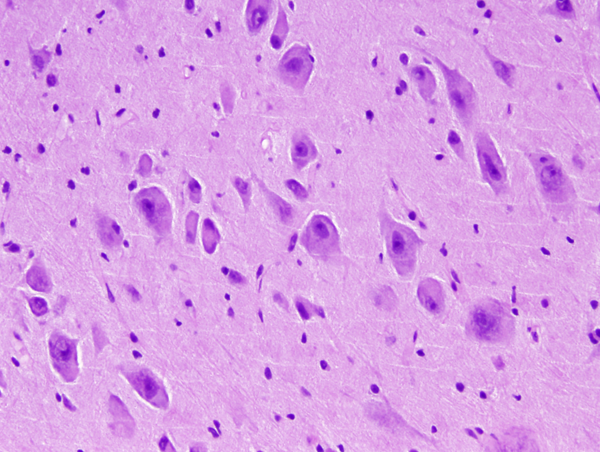 FCD type IIb- dysplasatic neurons |
 FCD type IIb-dysplastic neurons |
 FCD type IIb-balloon cells |
The most frequent pathology in brain tissue removed in epilepsy surgery in children is focal cortical dysplasia (FCD). Glioneuronal tumors (ganglioglioma, dysembryoblastic neuroepithelial tumor), vascular malformations, and other lesions are less frequent. A significant proportion have hippocampal sclerosis (HS), which is the most frequent lesion in older patients. FCD is a sporadic developmental malformation of the cerebral cortex that causes intractable seizures and cognitive impairment. The 2011 International League Against Epilepsy (ILEA) classification recognizes 3 types of FCD-see below.
FCD type I: abnormality of cortical layering (subdivided into Ia: abnormal vertical alignment of neurons; Ib: abnormal horizontal alignment; Ic: vertical and horizontal).
FCD type II: abnormal cortical architecture plus atypical cells (subdivided into IIa: dysmorphic neurons (DNs); and IIb DNs and balloon cells (BCs)).
FCD type III: cortical abnormality associated with other lesions (hippocampal sclerosis, glioneuronal tumors, vascular malformations, scars from old lesions of prenatal origin).
The core pathology of FCD is an abnormal cortical cytoarchitecture characterized by loss of normal layering. In FCD II, there are, in addition, abnormal cells of 2 kinds: DNs and BCs. The DNs are large or giant, dysmorphic pyramidal-like neurons with abnormal Nissl granules and cytoskeleton, and abnormal processes and orientation. The BCs have a large amount of glassy eosinophilic cytoplasm without processes and display ambiguous neuronal and glial features. Identical cells are seen in tubers of tuberous sclerosis complex (TSC). FCDIIa has DNs, and FCDIIb has DNs and BCs. Other findings include gliosis, loss of myelin, and ectopic neurons in the white matter.
Rapamycin is an antibiotic, antifungal and immunosuppressant . It is also a potent antagonist of mTOR (mammalian Target of Rapamycin), a serine/threonine kinase that plays a central role in multiple processes involved in brain development, including neuronal proliferation, neuronal growth, and axon formation. mTOR is regulated by several other proteins, including TSC1 and TSC2, which inhibit mTOR. Overactivatin of mTOR signaling (caused by mutations of TSC1 and TSC2 and other regulatory proteins) leads to cortical malformations, including tubers, FCDIIb, ganglioglioma, and hemimegalencephaly. The latter is a mosaic disorder with mTOR hyperactivation present in one hemisphere. mTOR hyperactivation is evident in pathological specimens from these disorders, which have been grouped under the umbrella term “mTORopathy”. The key features of mTORopathies are abnormal cortical cytoarchitecture, abnormal cells (DNs and BCs), intractable seizures, and cognitive abnormalities. mTOR overactivation has also been demonstrated in Neurofibromatosis 1, Sturge-Weber syndrome, Cowden syndrome, PMSE (Polyhydramnios, Megalencephaly, Symptomatic Epilepsy), and Proteus syndrome. Rapamycin has anti-epileptic action, and the rapamycin analog everolimus has been used successfully for the treatment of SEGA.
Further Reading
- Barkovich AJ, Kuzniecki RI Jackson GD, Guerrini R, and Dobyns WB. A developmental and genetic classification for malformations of cortical evelopment. Neurology 2005;65:1873-87 PubMed
- Guerini R, Dobyns WB, Barkovich AJ. Abnormal development of the human cerebral cortex: genetics, functional consequences and treatment options. Trends Neurosci 2008;31:154-62. PubMed
- Guerrini R, Parrini E. Neuronal migration disorders. Neurobiol Dis. 2010;38:154-66. PubMed
- Leventer RJ, Jansen A, Pilz DT, et al. Clinical and imaging heterogeneity of polymicrogyria: a study of 328 patients. Brain 2010;133:1415 – 27 PubMed
- Barkovich AJ. Current concepts of polymicrogyria. Neuroradiology 2010 52:479-87. PubMed
- Judkins AR, Martinez BS, Ferreira P, et al. Polymicrogyria Includes Fusion of th Molecular Layer and Decreased Neuronal Populations But Normal Cortical Laminar Organization. J Neuropathol Exp Neurol 2011;70:438-43. PubMed
- Crino PB. mTOR: A pathogenic signaling pathway in developmental brain malformations. Trends Mol Med 2011;17: 734-42. PubMed
- Aronica E, Becker AJ, Spreafico R. Malformations of Cortical Development. Brain Pathol 2012;22:380–401. PubMed
- Crino PB. mTOR: A pathogenic signaling pathway in developmental brain malformations. Trends Mol Med 2011;17: 734-42. PubMed
- Gaitanis JN, Donahue J. Focal Cortical Dysplasia. Pediatr Neurol 2013;49: 79-87. PubMed
- Parrini E, Conti V, Dobyns WB, Guerrini R. Genetic Basis of Brain Malformations. Mol Syndromol 2016;7:220-233. PubMed
Updated: October, 2017