Viral Diseases on the nervous system - general principles
Encephalitis is a clinical syndrome characterized by fever, altered consciousness, seizures, neurologic deficits and CSF pleocytosis. The cause of encephalitis is not known in about half of the cases. Among the cases with a known cause, about half are caused by viruses, the most common of which is Herpes Simplex Virus (HSV). Age-related or genetically-based defects of immunity confer susceptibility to certain types of viral encephalitis as do also environmental factors such as the season of the year and geographic location.
Viruses enter the body via the respiratory tract (mumps, measles), gastrointestinal tract (enteroviruses), by inoculation from insect bites (arthropod-borne viruses), and from animal bites (rabies). Most viruses reach the CNS via the bloodstream. Some viruses including herpes simplex virus(HSV), varicella-zoster virus (VZV), and rabies may also travel to the CNS along nerves. Viruses are obligate intracellular organisms. They use cellular machinery for their replication and damage or kill the cells they infect. Additional brain damage is caused by the cell-mediated immune reaction that they elicit. The cascade of events that begins with activation of T-lymphocytes by viruses includes the release of potent cytokines (INF-gamma, IL-2, TNF, lymphotoxin) and mobilization of macrophages that not only attack the viruses but assault the host, causing severe vascular and tissue injury.
Viral infection of the arachnoid membrane and CSF causes headache, stiff neck and mononuclear CSF pleocytosis with normal protein and glucose. This syndrome is called aseptic meningitis, because no bacterial organisms are isolated. The most common agents causing aseptic meningitis are enteroviruses. Involvement of neurons and glial cells by viruses (viral encephalitis) impairs neurological function and causes seizures, focal neurologic deficits, and coma. Encephalitis is accompanied by viral meningitis whereas aseptic meningitis may occur alone. The CNS is usually involved in the course of generalized viral infection. In babies (and with some viruses in adults also) there is often severe involvement of other organs. In many cases, however, damage of other organs is mild or absent. Some viruses have a predilection for certain groups of neurons. For instance, poliomyelitis attacks anterior horn cells and varicella-zoster involves sensory ganglion cells. However, even these viruses affect other neurons as well. Most viruses have no such special tropism.
 Microglial nodule. |
Histologically, viral infections show inflammation and brain damage. At an early phase inflammation includes neutrophils but later it consists predominantly of lymphocytes and macrophages. These cells infiltrate the arachnoid membrane and the brain diffusely but are more concentrated around blood vessels. Activation of microglia (indigenous macrophages of the brain) causes these cells to proliferate diffusely and form small clusters, microglial nodules which are a histological clue of viral infection. Tissue damage ranges from individual cell to diffuse brain necrosis involving large contiguous areas.
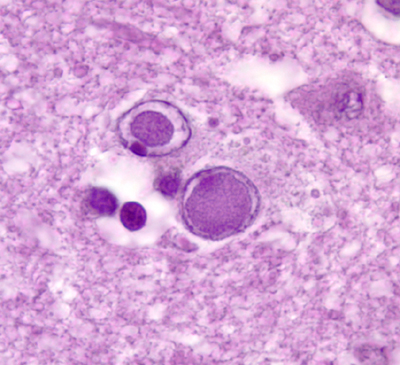 Intranuclear inclusions |
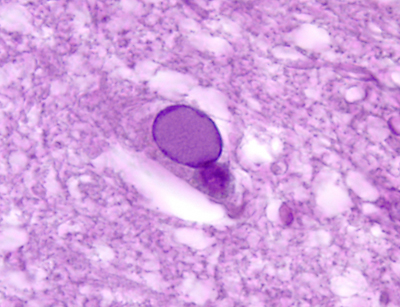 Intranuclear inclusion |
Certain viruses cause intranuclear and cytoplasmic inclusions.These are eosinophilic masses consisting of packed viral particles and products of their replication. Viruses that cause inclusions are herpes simplex, cytomegalovirus, varicella-zoster, papovaviruses, and measles. When the inflammatory and immune reactions of viral encephalitis subside, the lesions heal with glial scar formation. However, lost neurons do not regenerate. So, in the aftermath of viral infections, patients are often left with severe permanent neurological deficits.
Herpes simplex encephalitis
Adult and pediatric (post-neonatal) HSV encephalitis is caused most commonly by HSV type I. It is the most common year-round viral encephalitis. Most people become primarily infected with HSV in their teens or twenties. HSV type I is transmitted by the saliva. The initial infection causes a stomatitis. Following this, the virus remains latent in the trigeminal ganglion. From this location, reactivated virus can spread either to the skin, along the branches of the trigeminal nerve, causing sores on the lips (herpes labialis), or to the brain, infecting the meninges of the anterior and middle cranial fossae. From the meninges, the virus extends to the adjacent brain where it affects the temporal and inferior frontal lobes first and more severely, and then spreads to the rest of the brain. Adult HSV encephalitis is limited to the brain. Its symptoms are fever, confusion, coma, and seizures. In addition, because of the involvement of the frontal and temporal lobes, patients often display bizarre behaviour, personality changes, anosmia, and gustatory hallucinations. Survivors may have Korsakoff's amnesia, because of bilateral damage of the hippocampus, dementia, and seizures.
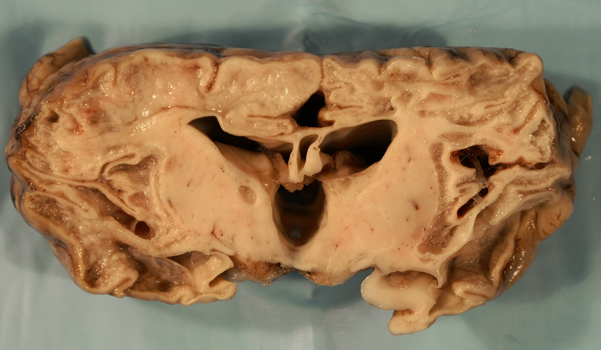
 Adult HSV encephalitis. |
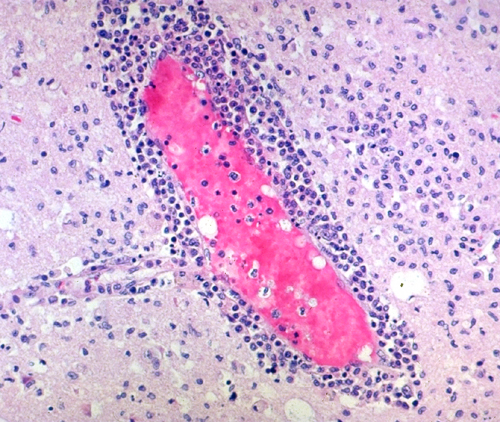 HSV encephalitis. |
The brain in advanced HSV encephalitis shows diffuse softening and edema, accentuated by hemorrhagic necrosis of the inferior frontal and temporal lobes. Microscopical examination in the acute phase shows meningeal and perivascular mononuclear cells, activated microglia, microglial nodules, and intranuclear inclusions. Without treatment, extensive necrosis, macrophage reaction and neovascularization develop. The end-stage is brain atrophy and gliosis. The degree of brain destruction (especially in the frontal and temporal lobes) and the inflammatory and reactive changes are more severe than any other viral encephalitis and can be detected by imaging. Atypical cases may have asymmetric pathology, mass-like necrotic and hemorrhagic fronto-temporal lesions, and a wider anatomical distribution, including myelitis and brainstem encephalitis. Some cases may be mild, causing only a mild reduction of the level of consciousness.
HSV type 2 causes similar disease and is also a frequent cause of aseptic meningitis. Both, HSV-1 and HSV-2 affect immunocompetent and immunosuppressed individuals. HSV-1 is more commmon in immunocompetent patiens and HSV-2 in immunosuppressed.
The CSF shows pleocytosis (early neutrophils, late lymphocytes) and elevated protein, depending on the degree of brain necrosis. Glucose is normal. HSV is hard to isolate from CSF. In the past, the diagnosis of HSV encephalitis required brain biopsy with fluorescent antibody staining using anti-HSV antibodies, microscopic study (to detect inclusions and viral particles) and viral culture. Now, HSV can be detected in CSF by polymerase chain reaction (PCR). HSV DNA may be detected by PCR years after the active infection. When a patient presents with fever, obtundation, seizures, and abnormal CSF, it is preferable to treat with antiviral agents (and antibiotics) rather than defer treatment until the diagnosis is established. In some cases, this approach will result in unnecessary use of antiviral agents. However, in patients with HSV encephalitis, prompt treatment saves lives and prevents permanent neurologic deficits.
Some children with HSV encephalitis develop an encephalopathy characterized by choreoathetosis weeks after CSF becomes negative and Acyclovir treatment is discontinued. A few adolescents and adults develop also psychiatric and other manifestations which appear either as a relapse or a continuation of the initial encephalitis. These patients have auto-antibodies against the NMDR receptor and other surface antigens, suggesting an auto-immune basis for this illness.
 Neonatal HSV encephalitis. |
Neonatal HSV encephalitis. About seventy percent of neonatal HSV infections are caused by HSV-2 (herpes genitalis) and thirty percent by HSV-1. HSV-1 is most commonly aquired during vaginal delivery by contact with secretions in the infected birth canal. Most infected mothers are asymptomatic. Cesarean delivery before rupture of membranes prevents these infections. In a few cases, HSV is transmitted across the placenta. A few babies also acquire the infection after birth, in the newborn nursery or at home. Untreated, neonatal HSV infection disseminates and involves the brain in most cases. The generalized infection affects the skin, eyes, liver, adrenal glands and other organs and presents clinically as neonatal sepsis. Encephalitis (lethargy progressing to coma, seizures, mononuclear CSF pleocytosis with elevated protein) develops one or two weeks after birth, sometimes later. A few cases of neonatal HSV encephalitis are mild but most cause a devastating diffuse necrotizing pathology, without predilection for the frontal and temporal lobes. In time, the lesions evolve into cystic encephalomalacia with microcephaly. The diagnosis is made by PCR of CSF. Treatment with Acyclovir significantly reduces mortality and improves the outcome in survivors.
Cytomegalovirus Encephalitis
Cytomegelovirus (CMV) encephalitis in adults is rare and usually occurs as part of a generalized CMV infection in immunocompromised patients.
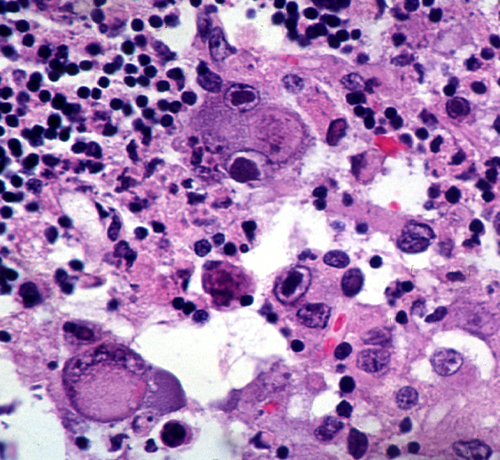 CMV encephalitis |
 Congenital CMV. |
 Congenital CMV |
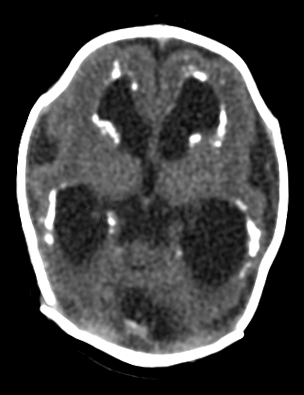 Congenital CMV. Head CT. |
Infected neurons and glial cells enlarge and develop cytoplasmic and intranuclear inclusions. Microglial nodules, lymphocytes, and macrophages infiltrate the lesions. Heavily infected areas become necrotic. The lesions have a predilection for the walls of the ventricles.
A pregnant mother who carries CMV may transmit the virus to the fetus, causing a generalized fetal CMV infection. This infection may develop at any stage during pregnancy and may continue after delivery. Infants with congenital CMV infection have variable involvement of the brain and other organs.
Prenatal CMV infection often causes necrosis of brain tissue, especially of the walls of the lateral ventricles. Necrotic areas calcify and can be detected by imaging. Infection before mid-gestation may derange the process of neuronal migration, causing microcephaly and cortical dysplasia. In some cases, fetal CMV infection destroys large parts of the brain, causing porencephaly, or schizencephaly. These lesions are thought to be caused by ischemia induced by the infection.
Enterovirus Infections
Enterovirus (EV) is a small RNA virus transmitted usually from human to human via fecal-oral or respiratory route. Humans are the only hosts. There are more than 100 serotypes of EV. Neonatal EV infection, usually transmitted from the mother, can cause fatal disseminated disease involving the brain, heart, liver, lungs, and other organs. In older children and adults, EV infection tends to be more limited and less severe and causes mucocutaneous eruptions, conjunctivitis, pleurodynia, respiratory illness, and myocarditis. Several EV serotypes affect also the CNS. Poliovirus, which causes poliomyelitis, is an EV. Polio has been eradicated in most of the world but still persists in some developing countries. Other (non polio) EV serotypes cause a wide spectrum of neurological disease. The most common and benign form of CNS involvement is aseptic meningitis. Less frequently, EV cause polio-like paralytic illness, transverse myelitis, Guillain-Barre-like disease, cranial neuropathy, and brainstem encephalitis with neurogenic pulmonary edema and shock. One of the most dangerous EV serotypes, EVA-71, which is also one of the agents of hand-foot-and-mouth disease, has caused large scale epidemics with severe neurological disease in Asia, Eastern Europe, and other regions. EV encephalitis has a predilection for the gray matter. Polio- is Greek for “gray”. The areas that are more severely involved are the gray matter of the spinal cord (accounting for the paralysis and subsequent denervation atrophy of muscle) and the gray matter of the brainstem (accounting for cranial neuropathies and probably also causing neurogenic pulmonary edema). Poliomyelitis affects also the motor cortex. Most EV are not strictly selective for motor neurons. The pathology consists of perivascular and meningeal inflammation (a mixture of neutrophils and mononuclear cells with the latter predominating in later stages), and microglial nodules. No intranuclear inclusions are seen. Neonatal EV encephalitis is disseminated and involves gray and white matter. The diagnosis is usually made by RTPCR of CSF or other samples.
The most common cause of viral meningoencephalitis in young infants is the picornavirus Human Parechovirus 3 (HPeV3),initially classified as EV and now reclassified as a separate genus.It causes cyclic outbreaks and nosocomial infections every 2-3 years, characterized by high fever, irritability, and seizures. There is no CSF pleiocytosis, and PCR is required for diagnosis. HPeV3 causes necrotic white matter lesions indistinguishable from periventricular leukomalacia.
Arbovirus Infections
Arthropod-borne viruses (arboviruses) are a group of small RNA viruses that are transmitted to humans by mosquito and other insect bites. The mosquitos acquire the virus by biting infected birds, rodents, and horses. Most arbovirus infections are asymptomatic but a minority can cause CNS infection including meningitis and severe or fatal encephalitis. The most common arboviruses causing neuroinvasive disease in North America are West Nile virus (WNV) and La Crosse Virus. Overall, arbovirus encephalitis is less common than encephalitis caused by enteroviruses or HSV. In recent years, Zika Virus (ZIKV) has emerged as an important cause of prenatal infection in South and Central America.
 WNV encephalitis |
 WNV encephalitis |
West Nile Virus encephalitis is the most common arbovirus encephalitis in adults. It occurs in 1 of 150 infected individuals, more frequently elderly patients, and presents with fever, followed, in a few days, by a change in mental status, which may progress to coma. Some patients with WNV encephalitis have severe asymmetric areflexic weakness or complete flaccid paralysis. Other neurological manifestations (ataxia, extrapyramidal, optic neuritis, seizures) are less common. The mortality rate in patients with encephalitis and muscle weakness is high, and survivors may have residual neurological deficits including fatigue, weakness, memory loss, and depression.Neuropathological examination shows changes of viral encephalomyelitis (perivascular lymphocytes, microglial nodules, neuronophagia, neuronal loss and focal demyelination and tissue necrosis), more severe in deep nuclei, brainstem, and spinal cord. The spinal cord pathology resembles poliomyelitis.
 La Crosse encephalitis |
 La Crosse encephalitis |
La Crosse encephalitis (LCE) is the most common arboviral disease in children. It occurs during the summer and fall and presents with fever, headache, vomiting, seizures, and disorientation. Hyponatremia and cerebral edema with herniations may develop. The CSF shows mononuclear pleocytosis with normal protein and glucose. RBCs may be present in some cases. The diagnosis is based on elevated serum IgM and IgG antibodies to La Crosse virus. LCE may mimic HSV encephalitis, and milder cases may be confused with enterovirus meningoencephalitis. LCE may be rarely fatal due to cerebral edema. About 12% of children surviving LCE are left with neurologic deficits. On microscopic examination, the brain shows perivascular mononuclear cells and microglial nodules.
Zika virus, an RNA flavivirus transmitted by Aedes egypti, was initially isolated in Uganda but has spread recently to other parts of the world, including South and Central America. It is most prevalent in Brazil, where hundreds of thousands of ZIKV infections have been reported. Most infections are asymptomatic. About 20% present with nonspecific symptoms that resemble dengue fever, including fever, headache, myalgia, arthralgia, and maculopapular rash. Rare infections cause the Guillain-Barre syndrome. When ZIKV infection occurs during pregnancy, the virus crosses the placenta and can affect the developing fetal brain with devastating effects. Babies born to ZIKV infected mothers have microcephaly. Prenatal and postnatal imaging studies and autopsies have shown severe microcephaly, brain atrophy, enlarged ventricles, abnormalities of cortical development including agyria, calcifications in the cortex, subcortical white matter, and in deep nuclei, abnormalities of the brainstem and cerebellum, abnormalities of the corpus callosum, and microphthalmia. ZIKV has been detected in the brain, CSF, placenta, and amniotic fluid of affected babies. The placenta in ZIKV infections is small and there is fetal growth retardation. No organ other than the brain is affected, suggesting that ZIKV is neurotropic. The pathology is somewhat similar to severe early prenatal CMV infection.
HIV Infections
Human immunodeficiency virus (HIV) infects and destroys selectively CD4 lymphocytes, causing immune deficiency. Consequently, AIDS patients are susceptible to opportunistic infections. The most common of these are CNS toxoplasmosis, cryptococcosis and other mycoses, CMV encephalitis, and papovavirus infections (progressive multifocal leukoencephalopathy). Historically, opportunistic infections attracted attention to AIDS long before it became known that HIV can also infect the brain. AIDS patients develop cerebral and other extranodal B-cell lymphomas due to loss of the surveillance function of T-cells.
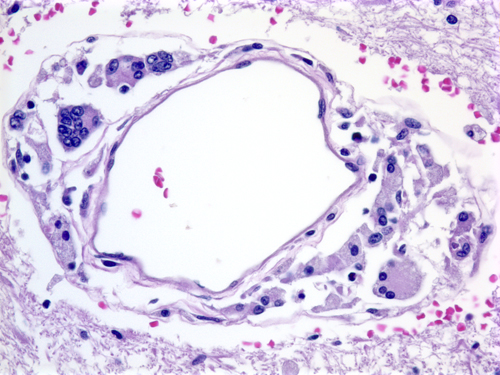 HIV encephalitis. Multinucleated giant cells. |
HIV also infects the nervous system directly, causing aseptic meningitis, encephalitis, leukoencephalopathy, myelopathy, neuropathy, and myopathy. The most common of these direct effects, HIV encephalitis (AIDS-dementia complex), causes progressive memory loss, intellectual deterioration, behavioral changes, and motor deficits. Pathologically, HIV encephalitis is characterized by diffuse myelin damage (spongy myelinopathy, gliosis), neuronal loss, vascular damage, microglial nodules, and lymphocytic infiltrates.The highest concentration of HIV is found in CNS macrophages. HIV envelope glycoproteins cause the membranes of HIV-infected macrophages to fuse, forming multinucleated giant cells (MGC) which are the hallmark of HIV encephalitis. The inflammatory reaction in HIV encephalitis is mild compared to other CNS infections because patients are immunodeficient. HIV can be transmitted from the infected mother to the fetus, resulting in congenital HIV infection. The lesions of congenital HIV encephalitis are more severe than those of the adult form and may result in microcephaly. One additional distinctive feature of congenital AIDS is basal ganglia calcification.
Restoration of the immune system of HIV patients following antiretroviral therapy often leads to new or exacerbated inflammation (Immune Reconstitution Inflammatory Syndrome-IRIS). The inflammatory reaction in IRIS may be directed against already present and previously tolerated agents such as JC virus or cryptococcus, components of HIV, or autoantigens. IRIS is usually systemic and only infrequently presents with neurological symptoms.
HIV is carried into the brain by infected CD4 lymphocytes and perivascular monocytes (see chapter one). Microglial cells pick up free viral particles through receptor-mediated endocytosis or aquire the virus by phagocytosing dying infected lymphocytes. The only cells that harbor HIV in the brain are perivascular monocytes and microglia. HIV has been detected by immunohistochemistry in neurons and astrocytes, but the significance of this observation is not clear. There is no productive infection of either neurons or glial cells. Therefore, brain damage is not due to a direct effect of HIV on either neurons or oligodendroglia, and neurological dysfunction does not correlate with viral load. Brain damage is caused mainly by cytokines (such as TNF) and neurotoxins (such as glutamate and NO) that are produced by activated monocytes and microglial cells. HIV envelope glycoprotein gp 120 also activates NMDA receptors and induces a cascade of neurotoxic events. The pathology of HIV myelopathy resembles subacute combined degeneration. This suggests that metabolic changes induced by HIV may contribute to the damage of myelin.
Further Reading
- French MA. Immune Reconstitution Inflammatory Syndrome: A Reappraisal. Clin Infect Dis 2009;48:101-7. PubMed
- Ramanathan S, Mohammad SS, Brilot F, Dale RC. Autoimmune encephalitis: Recent updates and emerging challenges. Journal of Clinical Neuroscience 21 (2014) 722–730 PubMed.
- Pardo CA, Vining EPG, Guo L, et al. The Pathology of Rasmussen Syndrome: Stages of Cortical Involvement and Neuropathological Studies in 45 Hemispherectomies. Epilepsia 2004;45:516-526. PubMed.
- Varadkar S, Bien CG, Kruse CA, et al. Rasmussen’s encephalitis: clinical features, pathobiology, and treatment advances. Lancet Neurol. 2014;13:195-205 PubMed.
- Mlakar J, Korva M, Tul N, et al. Zika Virus Associated with Microcephaly. N Engl J Med 2016;374:951-8.PubMed
- Soares de Oliveira-Szejnfeld P, Levine D, Melo AS, et al. Congenital Brain Abnormalities and Zika Virus: What the Radiologist Can Expect to See Prenatally and Postnatally. Radiology 2016;281:203-18.PubMed
- Dalmau J, Graus F. Antibody-Mediated Encephalitis. N Engl J Med 2018; 378:840-851PubMed
- Wiley CA. Emergent viral infections of the CNS. J Neuropathol Exp Neurol 2020;79:823-42
Updated: April 2023