HOLOPROSENCEPHALY
 Alobar holoprosencephaly |
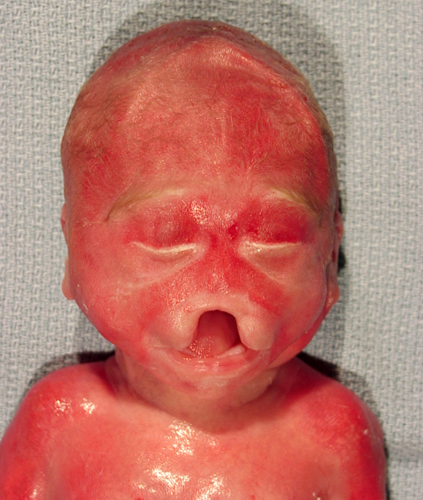 Facial abnormalities in HPE |
 Holoprosencephaly |
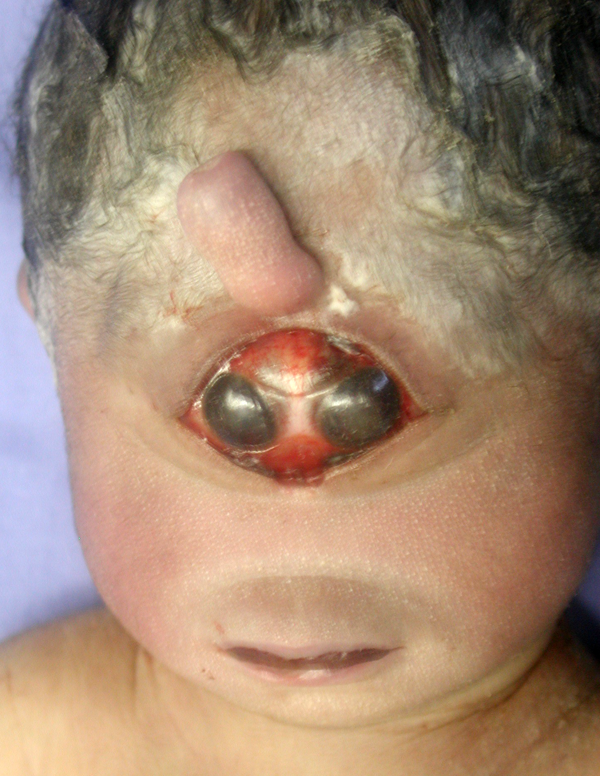 HPE |
Between the fourth to sixth week of gestation, the forebrain (prosencephalon-telencephalon) is divided into the two hemispheres. Absence of this cleavage results in a spectrum of malformations called holoprosencephaly (HPE). Because the olfactory nerves which are part of the rhinencephalon are absent, the term arrhinencephaly has also been applied to this malformation. However, in HPE, there is much more missing than the olfactory brain. In the most severe form, alobar HPE, the brain consists of a single spherical forebrain structure with a single ventricle. A large cyst which communicates with the ventricle is present in the posterior-dorsal part of the brain. The brain in alobar HPE is small and the gyral pattern and cortical architecture are abnormal. The eyes, which evaginate from the forebrain in the fourth week, are small and malformed or there is only one eye (cyclopia). Milder forms of HPE are semilobar HPE (fusion of frontal and parietal lobes with separation of the occipital lobes), lobar HPE (fusion of the ventral frontal lobes with separation of the rest of the hemispheres). The brainstem and cerebellum are preserved or show milder malformations. At the mild end of the spectrum is the middle interhemispheric fusion variant in which the anterior and posterior parts of the hemispheres are separate but the posterior frontal and parietal lobes along with the basal ganglia and thalamus are fused. Atelencephaly-aprosencephaly, a malformation in which the telencephalon is absent and only a nubbin of gliomesenchymal tissue with calcifications representing the diencephalon remains, has recently been added on the severe end of the spectrum.
A specrum of midline facial anomalies accompanies the brain malformations. In alobar HPE, these are severe and include a proboscis (a trunk-like structure above the single eye), a single nostril, cleft lip, and cleft palate. Milder brain pathology is accompanied by milder or subtle facial defects such as a central incisor and hypotelorism. The correlation between the facial anomalies and HPE was pointed out by DeMeyer in a paper titled "The face predicts the brain" (Pediatrics1964;34:256-63). Alobar HPE is incompatible with survival. Milder forms are associated with variable psychomotor retardation depending on the pathology. Diabetes insipidus is frequent in these patients.
HPE
is rare among live born infants
but very common in embryogenesis.
It has genetic and environmental
causes. Most cases are sporadic
but there are also autosomal
dominant, recessive, and X-linked
forms. HPE also occurs as a component
of multiple malformation syndromes
and in several chromosomal abnormalities,
most commonly trisomy 13, trisomy
18, and triploidy. Genetic HPE
is associated with four genes
and has been linked to seven
additional chromosomal loci.
The best known HPE gene is the Sonic
Hedgehog (SHH) gene
on 7q36 which is important for
ventral patterning of the forebrain.
Mutations of this gene cause
autosomal dominant HPE. Defective
cholesterol synthesis inhibits
SHH signaling resulting in HPE-like
malformations. Retinoic acid
participates in the SHH system. Excess
retinoic acid during
embryogenesis (from administration
of Accutane for acne) inhibits
SHH and causes HPE and other
malformations. The HPE-associated
gene TG-interacting factor (TGIF)
on 18p11, regulates retinoic
acid. Mutations of TGIF result
in unrestrained retinoic acid
activity and HPE. The multitude
of genes and chromosomal loci
associated with HPE underlines
the complexity of genetic programs
that are involved in embryonic
patterning and the intricate
interaction between genes and
environmental factors.
The
chemical messages that induce
the forebrain to divide into
two hemispheres, including SHH,
are first expressed in the prechordal
plate, an area rostral to the
notochord that gives rise to
the facial mesoderm. SHH is also
involved in craniofacial development.
AGENESIS OF THE CORPUS CALLOSUM
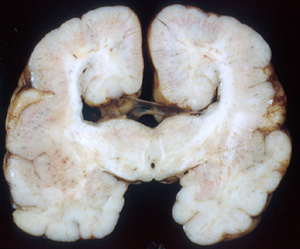 Agenesis of the corpus callosum |
At about 10 weeks of gestation, a glial bridge (massa commisuralis) forms between the two hemispheres, at the bottom of the interhemispheric fissure. Soon after this, axons begin to cross this bridge, forming the corpus callosum (CC). This process is completed by 18 to 20 weeks gestation. Agenesis of the corpus callosum (ACC) develops either if the bridge does not form or if axons fail to cross it. HPE can be mistaken for ACC.
ACC is one of the most common and probably the most genetically diverse brain malformation. It may occur as an isolated defect but is more frequently seen in association with other CNS and extraneural abnormalities. It is a component of more than 200 congenital syndromes. It occurs in several chromosomal abnormalities (including trisomy 13 and 18), malformation syndromes (including Aicardi, Apert, Fryns, Rubinstein-Taybi, and Smith-Lemli-Opitz syndrome), in association with CNS malformations (Dandy-Walker malformation, schizencephaly, polymicrogyria, lissencephaly-pachygyria), and in several inherited metabolic disorders (including the Zellweger syndrome, glycosylation disorders, nonketotic hyperglycinemia, pyruvate dehydrogenase deficiency, and respiratory chain abnormalities). Some patients with ACC have also extraneural malformations involving the skull, eyes, and ears, congenital heart disease, skeletal defects, and other malformations. ACC is associated with mutations of the L1 cell adhesion molecule, a cell surface glycoprotein that is important for guidance of migrating neurons (see neuronal migration defects).
ACC may be complete or partial, involving only the posterior part of the CC (splenium). Most patients have psychomotor retardation and about one third have seizures. A small minority-usually patients with isolated ACC-have normal neuromotor skills. When the CC is absent, the anterior horns of lateral ventricles have a bat-wing shape and the posterior horns are dilated and parallel to one another. The gap between the two hemispheres is filled sometimes by adipose tissue.
Further Reading
- Solomon BD, Rosenbaum KN, Meck JM, Muenke M. Holoprosencephaly due to numeric chromosome abnormalities. Am J Med Genet C Semin Med Genet. 2010 ;154C:146-8. PubMed
- Solomon BD, Mercier S, Vélez JI, et al.Analysis of genotype-phenotype correlations in human holoprosencephaly. Am J Med Genet C Semin Med Genet. 2010;154C:133-41. PubMed
- Roessler E, Muenke M.The molecular genetics of holoprosencephaly. Am J Med Genet C Semin Med Genet. 2010;154C:52-61. PubMed
- Romaniello R, Marelli S, Giorda R et al. Clinical Characterization, Genetics, and Long-Term Follow-up of a Large Cohort of Patients With Agenesis of the Corpus Callosum. J Child Neurol. 2016 Sep 28. pii: 0883073816664668. PubMed
- Fallet-Bianco C. Neuropathology of holoprosencephaly. Am J Med Genet C Semin Med Genet.2018 Jun;178(2):214-228. PubMed
Updated: March, 2023