HYDROCEPHALUS
Hydrocephalus is dilatation of the cerebral ventricles. This dilatation has a variety of causes, the common denominator of which is obstruction of CSF flow. Approximately 600-700 ml of CSF is produced daily by the choroid plexuses. From the lateral ventricles, CSF enters the third ventricle through the foramina of Monro and then flows into the fourth ventricle through the aqueduct. It exits from the fourth ventricle into the subarachnoid space through the foramina of Luschka and Magendie. It bathes the spinal cord and flows over the cerebral convexities to the arachnoid villi through which it is absorbed into the venous circulation. Hydrocephalus may result from the following causes:
Hypersecretion of CSF: choroid plexus papilloma Obstructive hydrocephalus
Defective filtration of CSF: postulated for low-pressure hydrocephalus. |
Hydrocephalus per se is not a malformation, but a deformation due to increased pressure in the ventricles. Many cases of hydrocephalus are caused by acquired lesions (tumors, subarachnoid hemorrhage, meningitis) some of which are included in the above list. One of the most common causes is intraventricular hemorrhage in premature infants. An even larger proportion of hydrocephalus cases are caused by developmental lesions or malformations many of which have a defined genetic basis. Some of these malformations have clear cut mechanical effects that explain the mechanism of hydrocephalus (see below). Some developmental forms of hydrocephalus are associated with other CNS and somatic malformations and defined syndromes. Many cases of developmental hydrocephalus are congenital but some develop later in life. The most common developmental forms of hydrocephalus are those that are associated with the Chiari II malformation and aqueductal obstruction. Developmental hydrocephalus includes also cases due to crowding of the posterior fossa (Chiari I, skeletal dysplasias), and other poorly cases of hydrocephalus without an apparent obstruction.
PATHOGENESIS OF BRAIN DAMAGE IN HYDROCEPHALUS
Initially, increasing pressure within the cerebral ventricles forces fluid through the ependymal lining into the periventricular white matter (transependymal edema). This is seen best on T2 MRI images. Pressure also causes the ventricles to dilate compressing brain tissue around them.
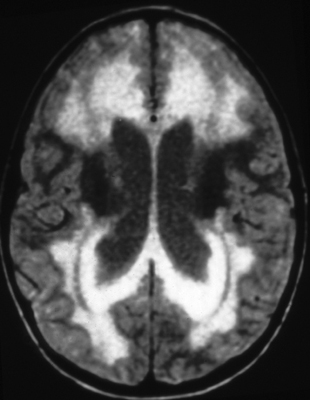 Transependymal edema |
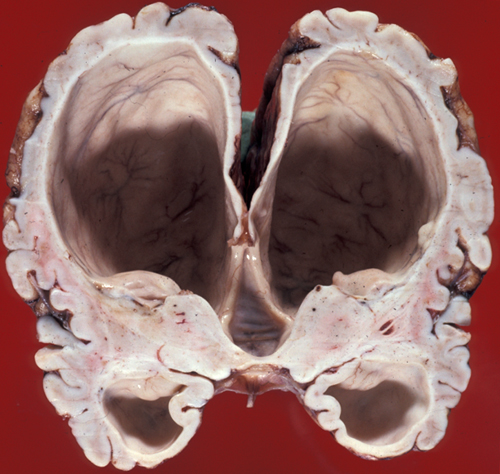 Hydrocephalus. White matter atrophy. |
The brunt of the damage falls mainly on the periventricular white matter which loses myelin and axons. Up to a certain point, the white matter changes are reversible, and often spectacular recovery is seen after shunting. If pressure is not relieved, permanent atrophy, first of the white matter and then of the cortex, develops. This causes spastic paralysis, loss of bladder function, and dementia. In severe hydrocephalus, the cortex and white matter may become paper thin and semitransparent such that the head transilluminates. Pressure cannot continue to build in the ventricles indefinitely. Something has to give or the patient will die from increased intracranial pressure. In young children, before the sutures close, the head may enlarge significantly. Sometimes the fibrosed subarachnoid space or a small aqueduct is forced open by increased pressure, thus relieving the obstruction. This is compensated hydrocephalus. Without relief, in extreme situations the cerebral mantle may break, releasing fluid into the subarachnoid space. Today, with prompt diagnosis and shunting, permanent neurological damage can be prevented in most cases.
AQUEDUCTAL ATRESIA AND STENOSIS
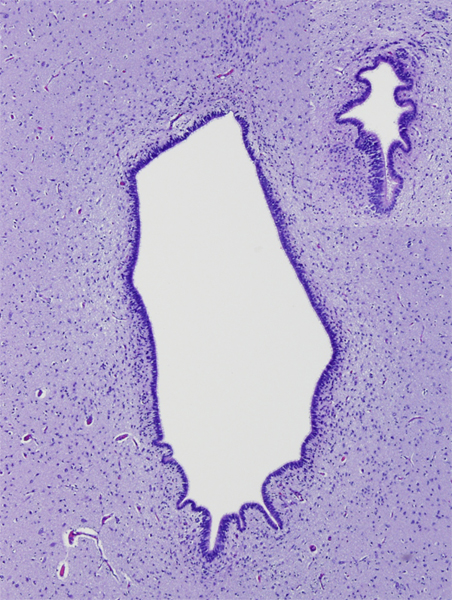 Aqueductal stenosis (upper right) |
 Aqueductal stenosis Hydrocephalus due to aqueductal stenosis. |
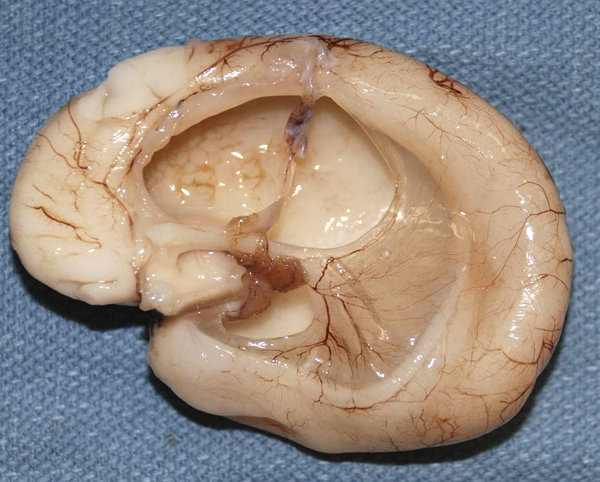 Aqueductal stenosis |
 Adducted thumbs |
 Absent pyramids |
Aqueductal atresia and aqueductal stenosis are the most common and severe causes of congenital hydrocephalus, with the Chiari II malformation (see below) being a close second. Aqueductal stenosis is pathologically distinct and it is a developmental lesion. It is a component of complex malformations and may be inherited in autosomal recessive or X-linked patterns. The best known form of aqueductal stenosis is X-linked aqueductal stenosis which occurs as part of the L1 syndrome.
The L1 syndrome encompasses the following 4 phenotypes:
- HSAS (X-linked hydrocephalus with stenosis of the aqueduct of Sylvius)
- MASA (mental retardation, adducted thumbs, shuffling gait, and aphasia)
- SPG1 (X-linked complicated hereditary spastic paraplegia type 1)
- X-linked complicated corpus callosum agenesis
 Aqueductal atresia |
Aqueductal atresia is a disruption that occurs in utero or post-natally. It may be caused by clots from intraventricular bleeding, infection, and other pathologies that cause gliosis and obliterate the aqueduct. Sometimes, a few rudimentary ependymal-lined tubules are seen in place of the aqueduct (aqueductal forking). These small channels are not enough to convey CSF from the third to the fourth ventricle. Aqueductal atresia is usually associated with other disruptive brain Aqueductal atresia cannot be distinguished by MRI from aqueductal stenosis, and the MRI diagnosis of aqueductal stenosis includes both entities.
CHIARI MALFORMATIONS
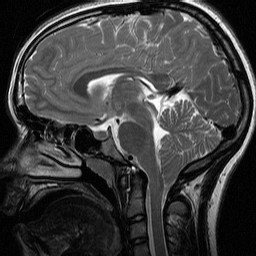 CM1 |
Five types of Chiari malformation (CM) haven described. The most common is Chiari malformation type 1 (CM1) and the most severe is Chiari malformation type 2 (CM2). In CM1 the volume of the posterior fossa is reduced leading to overcrowding and herniation of the cerebellar tonsils and dorsal cerebellum into the spinal canal. Many patients have syringomyelia and some have hydrocephalus. There is no neural tube defect. CM1 affects children and young adults and may be an incidental finding in imaging studies. Some patients are asymptomatic but others have headache, dizziness, cranial nerve abnormalities, spinal cord disturbances and other symptoms.
 CM2 |
CM2 is a syndrome or association of anomalies characterized by a) a neural tube defect, usually a lumbosacral myelomeningocele (MMC) b) abnormalities of the posterior fossa and craniocervical junction and c) hydrocephalus. The abnormality of the posterior fossa and its contents consists of a large foramen magnum, low insertion of the tentorium and a shallow posterior fossa. As a result of these deformities, the cerebellum and brainstem are crowded and displaced into the cervical canal. The cerebellum may also prolapse upward through the tentorial opening.
 CM2 |
 CM2 |
 CM2-Polygyria |
 CM2 |
The medulla is elongated and folded dorsally because the spinal cord is held in place by the dentate ligaments and cannot move down. The aqueduct and the fourth ventricle are collapsed. Often, there is aqueductal atresia. The foramina of Luschka lie in the spinal canal, and the subarachnoid space around them is collapsed and fibrotic. Blockage of CSF flow from these lesions causes hydrocephalus. Severe hydrocephalus causes parts of the cortex that had been hidden in the cerebral sulci to become externalized. The surface of the brain appears to have more gyri than normal and gives the false impression of polymicrogyria. However, unlike true polymicrogyria, the cortical cytoarchitecture is normal. The term polygyria describes more aptly the appearance of the cortex. Many CM2 patients also have hydromyelia or syringomyelia.
The key lesion in CM2 is probably the MMC, which is present in all CM2 cases. Leakage of CSF through the MMC creates low pressure in the spinal subarachnoid space that sucks the posterior fossa contents into the spinal canal. One consequence of this is collapse of the aqueduct and the 4th ventricle and blockage of the foramina of Lushka, resulting in hydrocephalus. Hydrocephalus is not a constant feature of CM2. It may not be present initially, and even later, it is present in in about 80% of cases. In that sense, the posterior fossa abnormality and hydrocephalus in CM2 represent a deformation sequence secondary to the MMC. However, in CM2 there are other cerebral and extracerebral malformations (notably agenesis of the corpus callosum in one third of cases). CM2 may also be associated with chromosomal abnormalities. These features suggest a more complex etiology and pathogenesis, combining malformation and mechanical deformation.
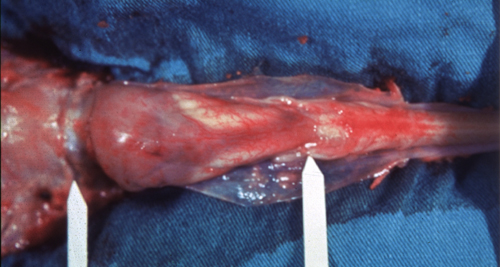
 Hydromyelia and amniotic contamination |
 Amniotic contamination |
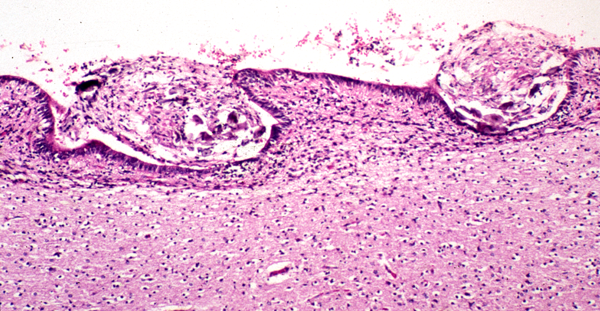 Amniotic contamination |
The respiratory abnormality that is frequently the cause of death, and cranial nerve abnormalities in CM2 are due to the brainstem pathology. The neuromotor deficit from the MMC is caused by the malformation of the spinal cord and is aggravated by subsequent damage of exposed neural tissue. Mechanical damage may occur during vaginal delivery. For this reason, cesarean section before the onset of labor and before rupture of the membranes is the preferred method of delivery of Chiari II babies. Chemical (and mechanical) damage may also occur from exposure of neural tissue to amniotic fluid and the squamous cells and lanugo hairs that float in it. This is prone to occur in open MMCs in which an amniotic-CSF fistula conveys amniotic products into the subarachnoid space, central canal, and, if the aqueduct is not blocked, even the cerebral ventricles. Amniotic squames and lanugo hairs congeal around the brainstem, penetrate superficially into neural tissue and cause significant mechanical and chemical irritation that adds to the already existing pathology. Early closure of the MMC by fetal surgery may prevent some of the posterior fossa changes and further protect neural tissue. Scarring at the site of the MMC may pull the spinal cord down leading to tethered cord which causes back pain, leg weakness and incontinence.
Hydrocephalus is also a frequent component of skeletal dysplasias and craniosynostosis syndromes in which there is a disparity between brain size and skull size. In some of these conditions, such as the ones caused by FGFR mutations, there is megalencephaly in addition to bony changes, accentuating the brain/skull disparity.
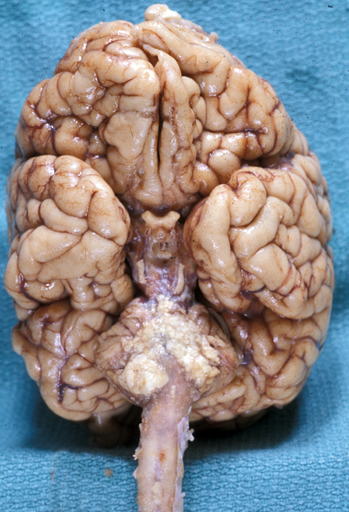
THE DANDY-WALKER MALFORMATION
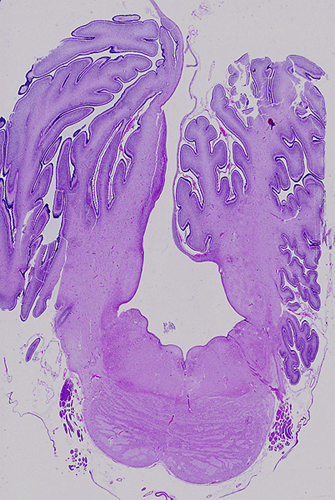 Dandy-Walker malformation |
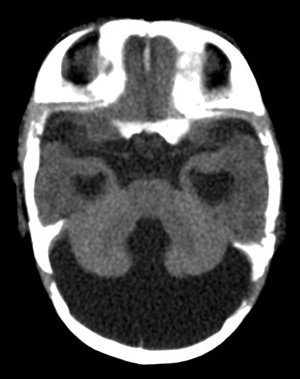 Dandy-Walker malformation |
The Dandy-Walker malformation (DWM) is a spectrum of posterior fossa abnormality (Dandy-Walker complex) the key feature of which is complete or partial agenesis of the cerebellar vermis. The cerebellar hemispheres are preserved and are joined by a thin membrane of neural tissue which forms the roof of the fourth ventricle. There is obstruction of CSF flow out of the fourth ventricle, the mechanism of which is not understood. As a result of this obstruction, the fourth ventricle dilates and the membrane that forms its roof balloons, creating a large posterior fossa cyst. This cyst pushes the tentorium upwards. Obstruction of CSF flow causes hydrocephalus. Milder variants of the DWM have lesser abnormalities.
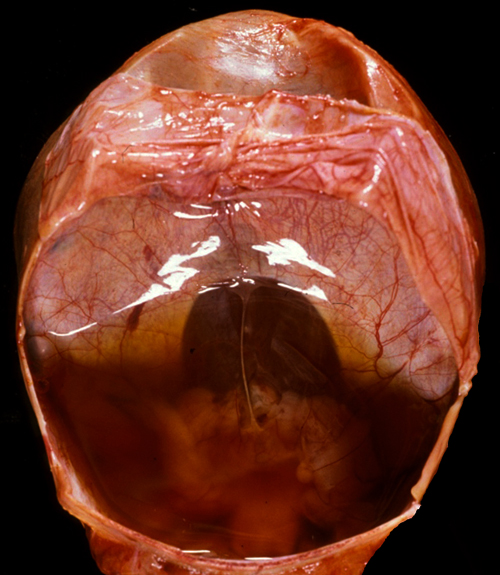 Dandy-Walker malformation |
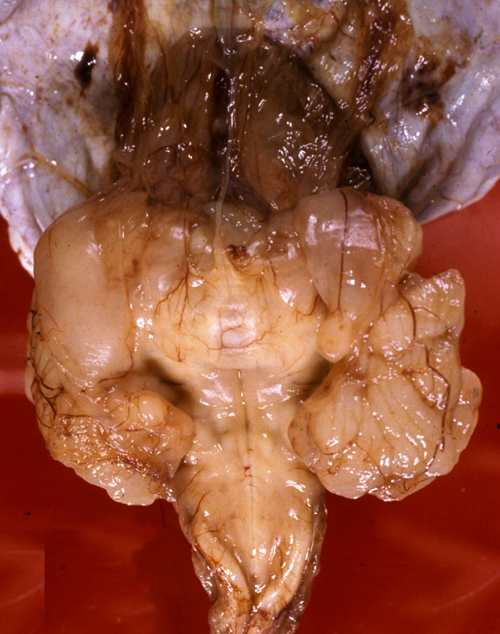 Dandy-Walker malformation Cerebellum from the DWM case on the left. The vermis is absent and the floor of the fourth ventricle (and floor of the posterior fossa cyst) is in plain view. A semitransparent membrane inserts along the margins of the fourth ventricle. The tentorial opening is seen in the upper part of the picture. |
The clinical profile, etiology, and genetics of the DWM are heterogeneous. Some patients have severe neurological deficits and additional developmental malformations of the brain (agenesis of the corpus callosum, neuronal migration defects) and of other organs. Some patients have relatively normal intelligence, especially after shunting. Most DWM cases are sporadic. There are rare familial cases associated with other malformation syndromes. the DWM has also been reported with trisomies of chromosomes 3, 9, 13, and 18. The DWM is very frequently diagnosed by prenatal ultrasound but, according to a recent study, in more than 50% of cases the ultrasound diagnosis is not suppported by the autopsy findings. Even using CT and MRI, the diagnosis of the DWM is difficult because in some planes of view it is difficult to distingish agenesis of the vermis from a large cisterna magna, cerebellar hypoplasia, or posterior fossa arachnoid cysts.
OTHER FORMS OF HYDROCEPHALUS
Hydrocephalus ex vacuo: dilatation of the cerebral ventricles due to loss of brain tissue. This is a common sequel of wasting brain diseases (leukodystrophies, multiple sclerosis, multiple strokes, Alzheimer's disease, Huntington's disease, etc.).
Idiopathic external hydrocephalus: a condition characterized by increased CSF volume and expansion of the subarachnoid space without ventricular dilatation, brain atrophy, intracranisl hypertension, or other pathology. This entity is common in infants and causes a large head and rapid growth of the head. It is not accompanied by neurological abnormality and usually resolves without treatment (benign macrocrania). It is probably due to immaturity of the arachnoid villi.
SYRINGOMYELIA
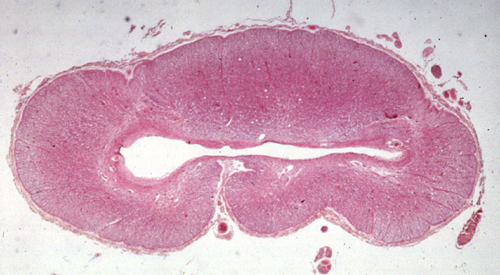 Syringomyelia |
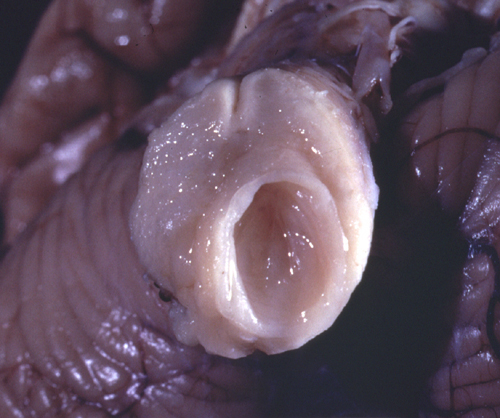 Syringobulbia |
Syringomyelia (syrinx, Gk a tubular cavity) is a tubular cavitation of the spinal cord which usually affects the cervical and upper thoracic segments. The cavity is in the central gray matter of the spinal cord. Initially it is separate from the central canal, but later, as it enlarges, it may communicate with it. Syringobulbia is an extension of the cavity from the spinal cord into the medulla. The syrinx is lined by glial tissue. It contains CSF-like fluid which accumulates progressively under pressure, causing atrophy of gray and white matter of the spinal cord. Symptoms from compression and atrophy of the spinal cord usually begin in the second or third decade of life. Initially there is dissociated anesthesia (segmental loss of pain and temperature sensation corresponding to the distribution of the syrinx with preservation of vibration and position sense), denervation atrophy of muscle, and kyphoscoliosis. Dissociated anesthesia is due to damage of the spinothalamic axons which cross anterior to the syrinx. As the cavity enlarges, more severe neurological damage may occur. Pressure may be relieved by shunting of the syrinx or by laminectomy.
Syringomyelia is often associated with the Chiari I malformation. In such cases, it has been postulated that obstruction of the foramina of Lushka which are displaced into the cervical canal keeps the central canal open. However, the syrinx is not actually the dilated central canal, and this mechanism does not apply to cases of syringomyelia without the Chiari I malformation or other craniocervical lesions. Thus, the pathogenesis of syringomyelia is unknown and is probably diverse. Syringomyelia is often seen above and below spinal tumors such as ependymoma, pilocytic astrocytoma, and hemangioblastoma. Some of these tumors tend to be cystic, and the syrinx may represent the cystic part of the tumor. Normally, the central canal closes postnatally, becoming a solid column of ependymal cells. Cystic dilatation of the central canal (hydromyelia) is a feature of the Chiari II malformation.
Further Reading
- Phillips JJ, Mahony BS, Siebert JR, et al. Dandy-Walker Malformation Complex. Correlation Between Ultrasonographic Diagnosis and Postmortem Neuropathology. Obstet Gynecol 2006;107:685-93. PubMed
- Adzick NS. Fetal surgery for spina bifida: Past, present, future. Sem Ped Surg 2013;22:10-17. PubMed
- Stevenson KL. Chiari Type II malformation: past, present, and future. Neurosurg Focus 2004;16:1-7. PubMed
- Tully HM, Ishak GE, Rue TC, et al. Two Hundred Thirty-Six Children With Developmental Hydrocephallus: Causes and Clinical Consequences. J Child Neurol 2016;31(3):309-20. PubMed
Updated: April, 2018