TUMORS OF GLIAL CELLS-A: ASTROCYTOMA, GLIOBLASTOMA, OLIGODENDROGLIOMA
The term "glioma" refers to all glial tumors in general (primarily glioblastoma, astrocytoma, oligodendroglioma, and ependymoma) but is also used sometimes instead of astrocytoma. The following introduction refers to astrocytic tumors in general, which are the most frequent gliomas.
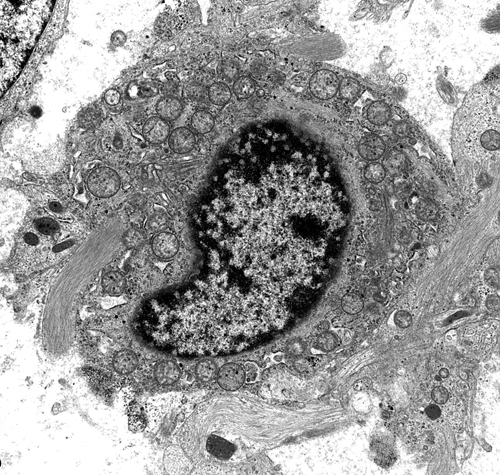 Cytoplasmic filaments |
 GFAP immunostain |
Neoplastic astrocytes share with normal ones the presence of intermediate cytoplasmic filaments. The protein of these filaments, glial fibrillary acidic protein (GFAP), can be detected by immunohistochemistry.
Astrocytomas have a wide spectrum of clinical behavior that can be expressed as a grade. The most commonly used WHO grading system uses four grades with grade I being the least malignant and grade IV the most malignant.
| Grade I-Pilocytic astrocytoma | Benign cytological features-see below |
| Grade II-Diffuse astrocytoma | Moderate cellularity-no anaplasia or mitotic activity |
| Grade III- Anaplastic astrocytoma | Cellularity, anaplasia, mitoses |
| Grade IV-Glioblastoma | Same as Grade III plus microvascular proliferation and necrosis |
"High-grade astrocytoma" includes WHO grades III and IV. Low-grade astrocytomas are more frequent in young patients and high-grade astrocytomas in older ones. Grading is used by oncologists to design treatment. It has practical usefulness and prognostic value but it may be subjective and some tumors do not fit neatly into a given grade. Progress in the molecular biology of gliomas has revealed some correlation between molecular changes and grade (see below). Grading is subject to sampling error, particularly with small (stereotactic needle) biopsies. Some astrocytic tumors are malignant from the outset. Others start as low-grade and evolve into high grade. The WHO system applies best to astrocytic tumors but, modified, it is used also for other gliomas. Adaptations of the WHO system are used to grade other brain tumors.
DIFFUSE ASTROCYTOMA (DA)- WHO GRADE II
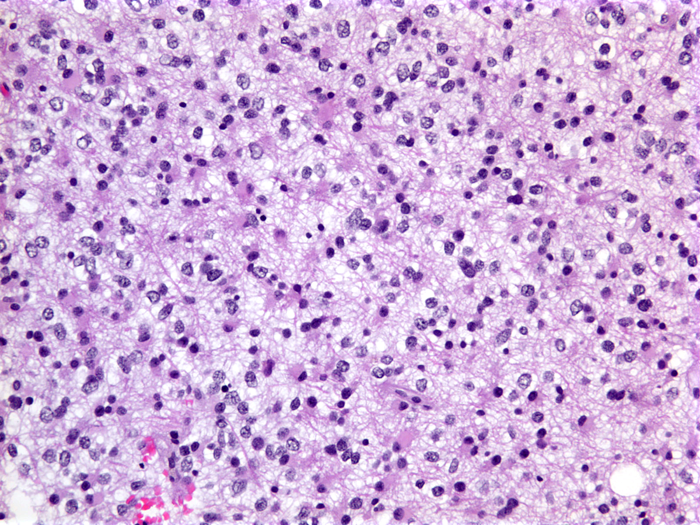 Diffuse astrocytoma |
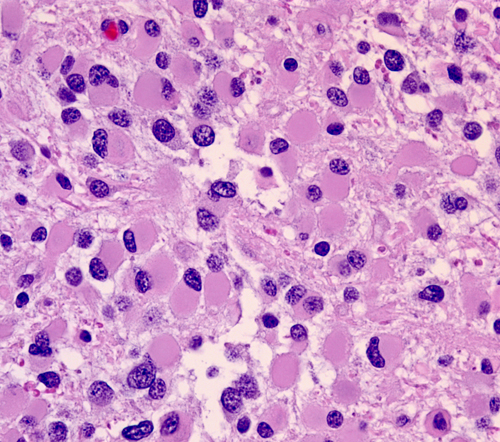 Gemistocytic astrocytoma |
Diffuse astrocytomas are most frequent in young adults. They arise anywhere in the CNS, but are most frequent in the cerebral hemispheres, especially frontal lobes. Most DAs are poorly demarcated and it is difficult to determine, by imaging, direct observation during surgery, or by gross pathological examination, where the tumor ends and normal tissue begins. All that is seen may be an enlargement of the involved portion of the brain and blurring of anatomical landmarks. Some DAs involve a large part of the brain in a diffuse fashion. Such cases have been called in the past gliomatosis cerebri. Histologically, the tumor cells can be stellate, spindle-shaped with fiber-like processes, or plump with a large eosinophilic cytoplasmic mass (gemistocytic astrocytoma). They show mild atypia and rare mitoses, and spread in a diffuse fashion but may also form microcysts and other tissue patterns. DAs cause seizures and focal deficits, corresponding to their location. They have an insidious presentation and grow slowly over a period of several years. Some remain histologically stable but many are gradually transformed over time into anaplastic astrocytoma and then glioblastoma.
ANAPLASTIC ASTROCYTOMA (AA)- WHO GRADE III
Anaplastic astrocytoma is clinically and pathologically an intermediate entity between Grade II DA and glioblastoma. In most respects it is similar to Grade II DA but is more cellular and has more prominent atypia and a higher mitotic rate and Ki67 proliferative index. Clinically also, AA is more rapidly evolving and has a shorter survival, commonly 3-5 years. Some AAs develop from pre-existing grade II DAs and others appear to arise de novo. AAs tend to transform into glioblastoma. Thus, some AAs seem to be a transitional stage between Grade II DA and glioblastoma.
GLIOMATOSIS CEREBRI
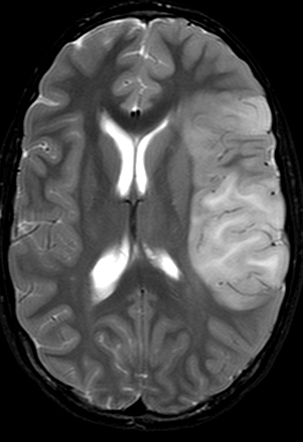 Gliomatosis |
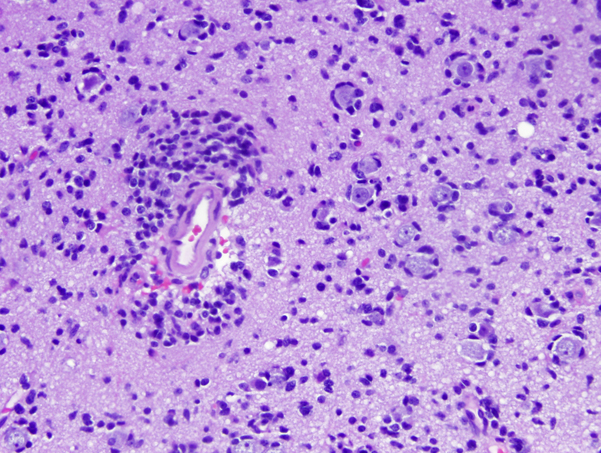 Gliomatosis |
Gliomatosis cerebri (GC) is a diffusely infiltrating neoplasm that may involve 2-3 lobes of the brain, an entire hemisphere, or the entire brain. Most cases of GC do not have a tumor mass, but in some a mass may be present from the start or appear later. Clinically, GC begins insidiously with changes of mental status, seizures, and focal deficits. This vague clinical presentation, coupled with a diffuse MRI signal abnormality, often suggests encephalitis, ADEM, or other entities. Histologically, most GC cases are composed of poorly differentiated glial cells, probably astrocytes, that infiltrate the brain diffusely and crowd around neurons and blood vessels and under the pia. These cases are comparable to WHO grade III astrocytoma. Such cases advance rapidly and have a poor prognosis. Some GC cases are due to proliferation of more mature astrocytes or oligodendrocytes and have a more protracted course.
GLIOBLASTOMA-WHO GRADE IV
Glioblastoma a.k.a. glioblastoma multiforme (GBM) is the most malignant glioma. About 18,000 patients are diagnosed with glioblastoma in the United States annually. It occurs most frequently in middle-aged adults. Its most common sites are the frontal and temporal lobes, but it may occur at any age and involve any part of the CNS. Glioblastoma arises most commonly de novo (primary glioblastoma). Some glioblastomas arise by malignant transformation of low-grade astrocytomas (secondary glioblastoma). Primary glioblastomas are more common in older patients and are more aggressive. Survival from glioblastoma rarely exceeds one year. Postoperative irradiation and chemotherapy prolong survival minimally.
Imaging shows a large irregular mass of variable density with cavitation, surrounded by a large area of edema. Vascularity accounts for the contrast-enhancing properties of glioblastoma but contrast enhancement should not be equated with malignancy; pilocytic astrocytoma also enhances.
 Glioblastoma |
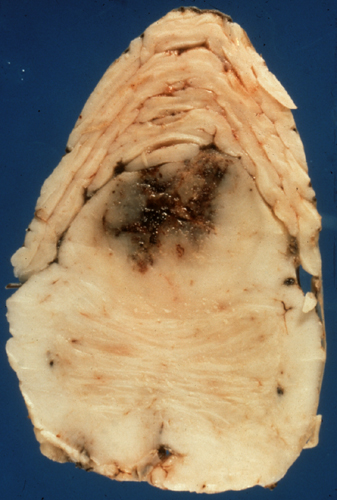 GBM of the pons |
On naked eye examination, glioblastoma is a poorly defined intra-axial mass with variegated (multiform) appearance due to necrosis and hemorrhage. If the tumor is near the center of the cerebrum, it may spread from one hemisphere to the other across the corpus callosum. Other malignant BT can have the same pattern. Also, large MS lesions, especially Schilder's disease, may involve both hemispheres and be confused with glioblastoma.
Microscopically, glioblastoma shows high cellularity, cellular and nuclear anaplasia which is the basis of the designation "multiforme", mitoses, microvascular proliferation, and necrosis. Glioblastoma has a wide range of histological appearances. Small cell glioblastoma is composed of poorly differentiated, uniform, small cells. At the other end of the spectrum, giant cell glioblastoma is characterized by extreme anaplasia. This rare variant tends to be superficial and more circumscribed.
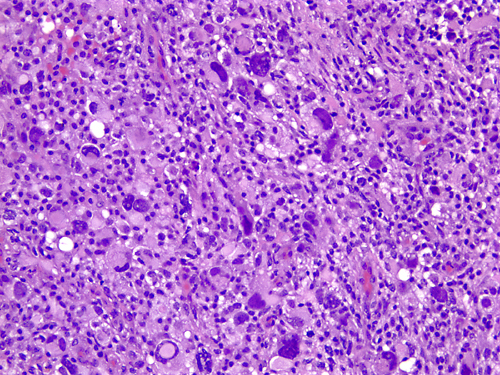 Anaplasia |
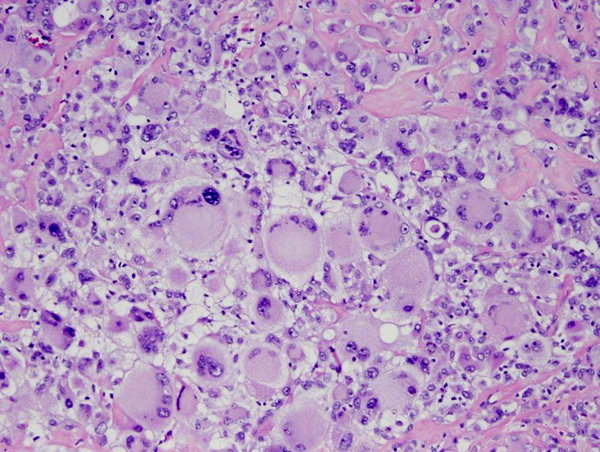 Giant cell glioblastoma |
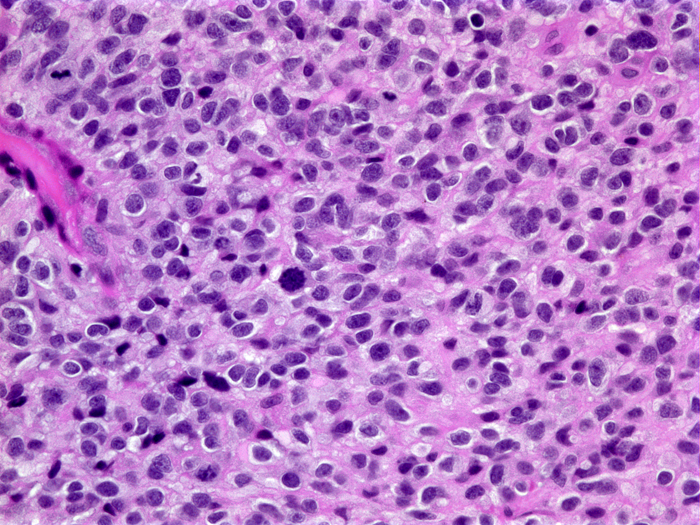 Small cell glioblastoma |
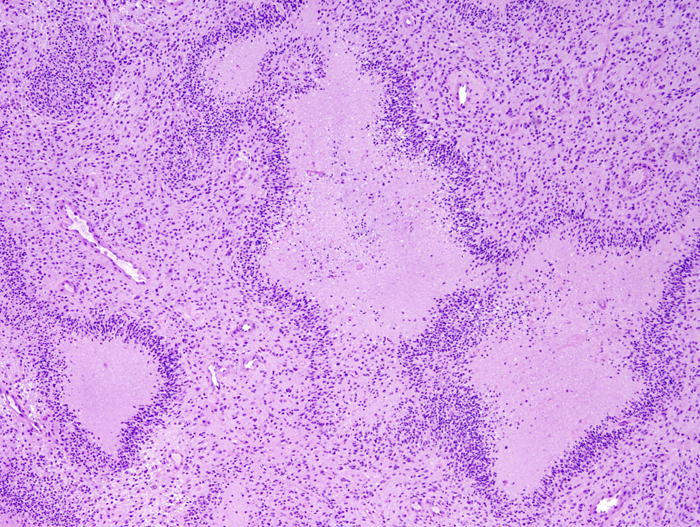 Necrosis and pseudopalisading |
 Microvascular proliferation |
Densely cellular arrays of tumor cells are often arranged in a perpendicular (pseudopalisading) fashion around serpiginous necrotic areas. It has been proposed that these tumor cells are migrating away from a central hypoxic area. Thrombosed vessels are often seen in the central necrotic area while microvascular proliferation in adjacent areas sustains tumor growth. Glioblastoma is one of the most highly vascular solid tumors. Angiogenesis in glioblastoma is a complex molecular process. Hypoxia, which develops as glioblastoma outgrows its vascular supply, induces upregulation of hypoxia inducible factor 1 (HIF-1), which, in turn, stimulates the expression of vascular endothelial growth factor (VEGF). Overexpression of these genes in glioblastoma induces formation of new vessels, which allow continuing tumor growth. The new vessels are often arranged in glomeruloid formations, and lack a blood-brain barrier. The latter property contributes to cerebral edema, a clinically important feature of glioblastoma. Primary glioblastomas are often composed of small undifferentiated cells (small cell glioblastoma) and show extensive ischemic necrosis and a higher proliferative index. Secondary glioblastomas are composed of larger cells with astrocytic differentiation.
MOLECULAR CHANGES IN ASTROCYTOMA-GLIOBLASTOMA
Isocitrate dehydrogenase (IDH)
DA, glioblastoma, and oligodendroglioma are genetically related tumors and share mutations of isocitrate dehydrogenase (IDH), a citric acid cycle enzyme that catalyzes the conversion of isocitrate to α-ketoglutarate (α-KG), producing NADPH. IDH1 is mutated in 70-80% of grade II and III astrocytomas, oligodendrogliomas, oligoastrocytomas, and in secondary but not primary glioblastomas. IDH2 mutations are more common in oligodendrogliomas. Tumor cells with IDH mutations lose the ability to produce α-KG and gain the ability to produce 2-hydroxyglutarate (2-HG), an α-KG antagonist. These metabolic disruptions reduce anti-oxidant function and cause histone and DNA methylation, which impairs stem cell differentiation, leading to tumor formation.
Beyond IDH mutations, the genetic lines of astrocytic and oligodendroglial tumors diverge. One subset of IDH1 mutant gliomas develops TP53 mutations and ATRX loss (see below). These tumors evolve into DA, AA, and glioblastoma. Another subset has 1p19q loss and evolves into oligodendroglioma.
Patients with brain tumors and mutant IDH1 have longer survival than patients with wild-type IDH1. IDH mutations may make tumor cells less viable by increasing their susceptibility to oxidative damage. IDH1 mutations are not seen in diffuse astrocytomas of children, and are not a feature of pilocytic astrocytoma or pleomorphic xanthoastrocytoma. IDH mutations can be detected by immunohistochemistry, which shows cytoplasmic and weaker nuclear staining for IDH. Their detection is an important tool in the diagnosis of gliomas.
Tumor Protein 53 (TP53) is a tumor suppressor encoded by the TP53 gene on 17p. It repairs DNA damage and induces apoptosis when damage cannot be repaired. Its mutation promotes tumor formation by enabling damaged cells to survive and grow. TP53 mutations are common in diffuse astrocytoma, anaplastic astrocytoma, and secondary glioblastoma, and help distinguish glioma from gliosis. Mutations can be detected by immunohistochemistry, which shows strong nuclear staining for p53.
ATRX (α-thalassemia/mental retardation syndrome X-linked)
The ATRX gene, on Xq21.1, is important for chromatin remodeling, and, through this process, regulates the activity of other genes. ATRX mutations have been reported in a large proportion of adult grade II and grade III astrocytomas, oligoastrocytomas, and secondary glioblastomas. Mutations are less frequent in oligodendrogliomas and pediatric glioblastomas, and rare in primary glioblastomas. These mutations result in loss of nuclear ATRX staining which can be detected by immunohistochemistry.
The TERT (telomerase reverse transcriptase) gene, located on 5p15, encodes the catalytic subunit of telomerase, which maintains telomeres. Mutations not of the protein-coding sequence but of the TERT promoter were first reported in melanomas and subsequently in several other tumors. TERT promoter mutations are also very common in brain tumors, especially primary glioblastomas, oligodendrogliomas, some medulloblastomas, and other brain tumors.
PTEN and LOH chromosome 10
Loss of the tumor suppressor gene PTEN (phosphatase and tensin homolog deleted on chromosome 10) on 10q as a result of deletion of 10q or the entire chromosome 10 is the most common genetic abnormality in glioblastoma, occurring in the majority of these tumors and less frequently in grade II and III astrocytomas.
Epidermal Growth Factor Receptor (EGFR)
Overexpression of EGFR, on 7p, occurs in 40% of glioblastomas (more commonly primary ones) and less frequently in lower-grade astrocytomas and provides a potential target for EGFR inhibitors.
MGMT
An important epigenetic alteration in glioblastoma and other gliomas is silencing of the O6-methylguanine-DNA methyltransferase gene (MGMT) through hypermethylation of its promoter. The MGMT gene encodes a DNA repair enzyme which repairs DNA crosslinks created by alkylating agents, such as temozolomide (TMZ), thus countering the effects of chemotherapy. Its inactivation makes these tumors more sensitive to TMZ. MGMT promoter methylation is detected by promoter methylation assay and is found in a large proportion of diffuse gliomas, including glioblastomas. Glioblastoma patients with hypermethylated MGMT treated with TMZ and radiotherapy survive longer than similarly treated patients without MGMT hypermethylation.
Detection of many of the molecular changes that underlie the development of gliomas and other brain tumors can now be done by immunohistochemistry and is entering the mainstream of the diagnostic workup of brain tumors. The results can be used for grading and customizing management by inhibiting activated pathways. The labeling index, determined by Ki-67 (MIB-1) immunohistochemistry, can help distinguish grade II from grade III astrocytoma. Even low-grade astrocytomas may be clinically malignant because their location and diffuse spread make surgical excision impossible, and they are not very susceptible to chemotherapy or radiation.
OLIGODENDROGLIOMA AND OLIGOASTROCYTOMA
Oligodendroglioma |
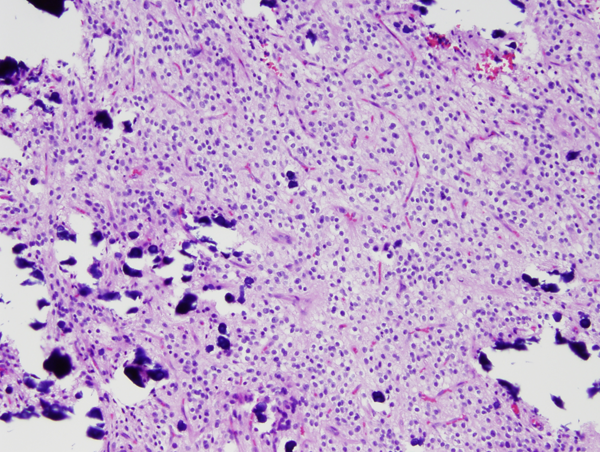 Oligodendroglioma |
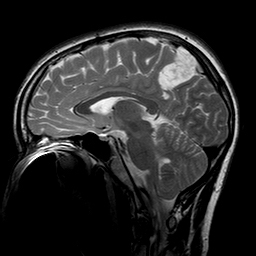 Oligodendroglioma MRI |
Oligodendrogliomas are 5%-6% of gliomas. They are insidious, slowly growing tumors and arise usually in the cerebral hemispheres of middle-aged adults. Oligodendrogliomas are more circumscribed than astrocytomas. Microscopically, the tumor cells are uniform and have round central nuclei with fine chromatin surrounded by a clear halo (unstained cytoplasm), which is an artefact of processing. Some tumors contain mini-gemistocytes. They infiltrate the cortex diffusely and accumulate under the pia, around neurons (satellitosis), and around blood vessels. Oligodendrogliomas are traversed by a delicate capillary network and have a tendency to calcify, which is helpful in radiological and histological diagnosis. They may form microcysts. On EM examination, the tumor cells produce abundant plasma membrane that tends to form concentric layers mimicking myelin. Some oligodendrogliomas contain neoplastic astrocytes which are mixed with the oligodendroglial cells or grow in adjacent but separate areas. Such mixed tumors are called oligoastrocytomas. Oligodendrogliomas and oligoastrocytomas can be classified as low-grade (WHO grade II) or high-grade/anaplastic (WHO grade III) based on cellularity, anaplasia, mitotic activity, microvascular proliferation, and necrosis. Median survival for grade II and grade III oligodendroglioma is about 11 and 4–5 years, respectively; survival is shorter for oligoastrocytomas.
The signature molecular change of oligodendroglioma is co-deletion of the entire arms of 1p and 19q, caused by an unbalanced translocation t(1;19)(q10;p10). In addition to being a diagnostic marker for oligodendroglioma, the 1p19q deletion predicts increased chemosensitivity and better prognosis, and is associated with classic oligodendroglioma morphology and frontal location. The 1p19q codeletion is specific for oligodendroglioma. The vast majority of oligodendrogliomas with the 1p19q codeletion also carry IDH1 mutations. Most pediatric oligodendrogliomas lack the 1p19q deletion and IDH mutation.
Further Reading
- Mellinghoff IK et al. Molecular Determinants of the Response of Glioblastomas to EGFR Kinase Inhibitors. N Engl J Med 2005;353:2012-24. PubMed
- Hegi ME et al. Clinical trial substantiates the predictive value of O-6-methylguanine-DNA methyltransferase promoter methylation in glioblastoma patients treated with temozolomide. Clin Cancer Res. 2004 Apr 15;10(8):1871-4. PubMed
- Rong Y, Durden DL, Van Meir EG, Brat DJ. "Pseudopalisading" necrosis in glioblastoma: a familiar morphologic feature that links vascular pathology, hypoxia, and angiogenesis. J Neuropathol Exp Neurol 2006;65:529-39. PubMed
- Schiff D, Brown PD, Giannini C. Outcome of adult low-grade glioma. The impact of prognostic factors and treatment. Neurology 2007;69:1366-73. PubMed
- Rodriguez FG, Giannini C. Oligodendroglial tumors: diagnostic and molecular pathology. Seminars in Diagnostic Pathology 2010;27:136-145.PubMed
- Ichimura K. Molecular pathogenesis of IDH mutations in gliomas. Brain Tumor Pathol 2012 Mar 8. [Epub ahead of print] PubMed
- Eckel-Passow JE et al. Glioma Groups Based on 1p/19q, IDH, and TERT Promoter Mutations in Tumors. New Engl J Med 2015;372:2499-2508.PubMed
- The Cancer Genome Atlas Research Network. Comprehensive, integrative genomic analysis of diffuse lower-grade gliomas. New Engl J Med 2015;372:2481-2498.PubMed
- Wesseling P, van den Bent M, Perry A. Oligodendroglioma: pathology, molecular mechanisms and markers. Acta Neuropathol 2015;129:809-27.PubMed
- Appin CL, Brat DJ. Biomarker-driven diagnosis. Mol Aspects Med. 2015;45:87-96. PubMed
- Louis DN, Perry A, Reifenberger G, et al. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathol 2016;131:803-20. PubMed
- Tanboon J, Williams EA, Louis DN. The Use of Immunohistochemical Surrogates for Signature Molecular Alterations in Glioma. J Neuropathol Exp Neurol 2016; 75:4-18. PubMed
