WHITE MATTER INJURY
White matter injury (WMI) is a form of ischemic white matter pathology which affects premature infants especially ones with cardiorespiratory abnormalities and sepsis. Mature infants, especially those with congenital heart disease, may also be affected. WMI and germinal matrix-intraventricular hemorrhage (see further on) are largely responsible for the cerebral palsy (CP), cognitive impairment and other neurological impairments that so frequently affect preterm infants. WMI usually develops in the neonatal period but may also occur in utero. There are 3 "grades" of WMI: focal cystic necrosis, focal microscopic necrosis, and nonnecrotic, diffuse white matter injury (DWMI). Today, thanks to improved neonatal intensive care, focal cystic necrosis is infrequent. The most common pattern is DWMI.
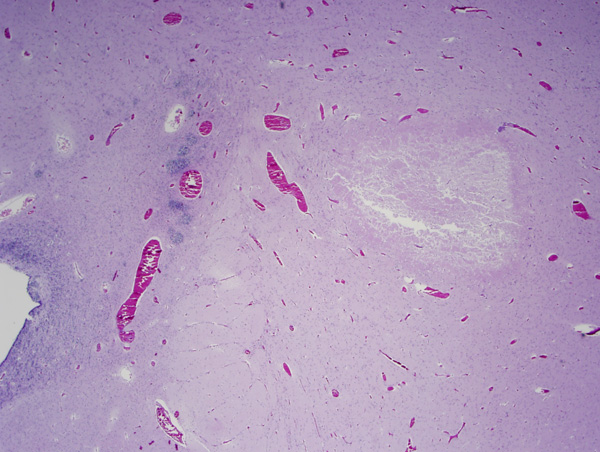 Focal cystic necrosis |
 Focal cystic necrosis Axonal swellings |
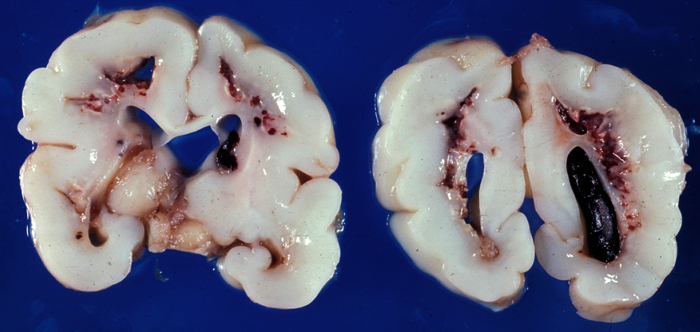 Focal cystic necrosisL |
 Focal cystic necrosis-Calcified lesions |
Focal cystic necrosis, was originally described in autopsy studies as periventricular leukomalacia (PVL) and in some older papers, the term PVL encompasses all forms of WMI. It is the most severe form of WMI affecting about 5% of patients and is characterized by bilateral, roughly symmetric foci of white matter necrosis around the lateral ventricles, especially in the frontal and occipital lobes. The evolution of these lesions is similar to infarcts, i.e. liquefaction, phagocytosis, cavitation, and gliosis. Axonal damage is evident by the presence of axonal swellings, which may be obvious on H&E stains and can be detected also with immunostains for Beta Amyloid Precursor Protein. An added feature of focal cystic necrosis and other necrotic brain lesions in fetuses and neonates is calcification, which makes lesions appear as whitish spots in the periventricular white matter. Large necrotic lesions cavitate in 2-4 weeks and remain cystic.
 White matter gliosis |
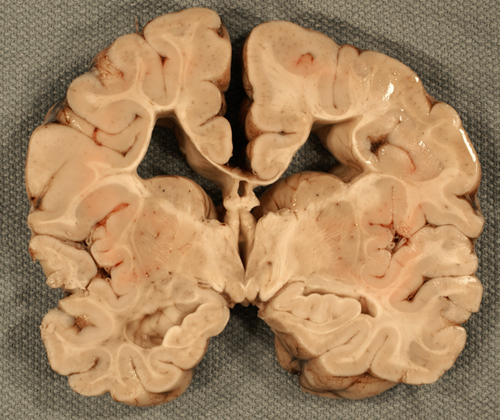 White matter injury |
In focal microscopic necrosis, small necrotic lesions form microcysts or do not cavitate at all but collapse into glial scars which can be detected as punctate lesions on MRI. DWMI does not have focal lesions but is characterized by hypomyelination, gliosis, decreased white matter mass, dilatation of the lateral ventricles (ventriculomegaly), and atrophy of the corpus callosum. Cortical ischemic lesions and loss of cortical and thalamic neurons are present in some cases.
 Focal cystic necrosis-cranial ultrasound |
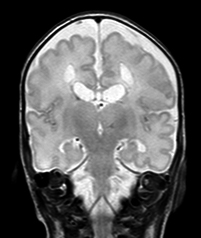 Focal cystic necrosis.-MRI |
The method of choice for the diagnosis of focal cystic necrosis is cranial ultrasound (US). The recommendation for infants less than 30 weeks’ gestation is a screening ultrasound at 7-14 days and a repeat ultrasound at 30-40 weeks. Echodensities correlate with the acute phase of necrosis. They transition into coalescent echolucencies (cavities) that give the affected white matter a “Swiss cheese” appearance.
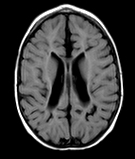 DWMI MRI of advanced lesion |
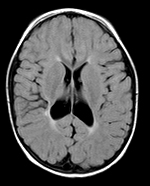 DWM MRI of advanced lesion |
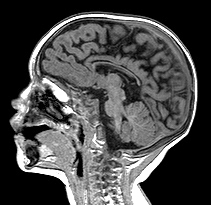 DWMI MRI of advanced lesion |
The MRI is more accurate than US for the detection of focal microscopic necrosis and DWMI and for following-up of acute lesions detected earlier by US. The MRI findings in DWMI are 1) loss of white matter with atrophy of the corpus callosum; 2) enlargement of the ventricles with an irregular angular (scalloped) appearance of their contours; and 3) abnormal or delayed myelination.
Congenital heart disease (CHD) is the most common congenital abnormality, affecting about 8 of every 1000 newborn infants. The fetal circulation is designed in a way that conveys a large proportion of oxygenated blood preferentially to the brain. In some forms of severe CHD, especially transposition of great vessels and hypoplastic left heart syndrome, the brain develops in a state of chronic ischemia and hypoxia. A large proportion of such patients have motor, cognitive, and behavioral abnormalities. The encephalopathy of CHD affects mature infants and has identical imaging and pathological features to DWMI affecting preterm infants. In patients with complex CHD, DWMI is caused by cerebral ischemia and hypoxia which occur prenatally or after birth, preoperatively or postoperatively, especially following deep hypothermic circulatory arrest and cardiopulmonary bypass, procedures that are necessary for its surgical correction. Moreover, chronic hypoxia in complex CHD affects neuronal migration and cortical maturation such that patients have smaller brains with decreased frontal lobe gyration.
WMI is caused by hypoxia/ischemia and by infection. In focal cystic necrosis, ischemia, causes white matter necrosis (infarction) that affects all tissue elements, including axons and oligodendrocytes. The lesions are located at the termination of major cerebral vessels in the border zones between the ACA, MCA, and PCA and involve the deeper parts of the white matter, where the developing vascular tree has not yet advanced. Hypoxia causes activation of microglia, the tissue histiocyte in the CNS, leading to secretion of toxic oxygen and nitrogen radicals and the release of glutamate. Free radicals and glutamate are the key agents of cellular injury in PVL and other forms of HIE (see alsoAsphyxia
and HIE in Mature Infants).
Inflammatory cytokines and activated monocytes that are generated during maternal, placental, and fetal infection enter the brain by crossing the immature blood-brain barrier and activate microglial cells setting in motion the same toxic cascade that damages the white matter in ischemia. Clinical studies show a high association of PVL and CP with chorioamnionitis, funisitis and premature rupture of membranes.
DWMI is caused by selective damage of premyelinating oligodendrocytes (pre-OL). In the premature brain, there is no myelin. The white matter is populated by oligodendrocyte (OL) precursors that evolve in 3 stages: OL progenitors, pre-OLs, and immature OLs. The principal targets of oxygen and nitrogen radicals and of glutamate are pre-OLs, which are vulnerable to hypoxia because they are deficient in Superoxide Dismutase, the key antioxidant enzyme. Additionally, the premature white matter is rich in iron, the most important source of free radicals. So, the key event in DWMI is loss of pre-OLs and the main outcome is deficient myelination.
The direct effects of the structural lesions of WMI are compounded by dysmaturation of the brain. Some of this dysmaturation is caused by the white matter lesions. WMI, especially during the second trimester, may affect the subplate (an ephemeral neuronal layer under the permanent cortex) and late migrating neurons that traverse the white matter on their way to the deep cortical layers. The subplate is important for the development of connections between the thalamus and cortex. Its premature loss disconnects the cortex from the thalamus. Damage of migrating neurons results in decreased cortical volume and decreased cortical folding. Neuronal loss in WMI adds to the devastating effects of myelin and axonal damage. There is also a suggestion that prematurity alone, even without WMI, adversely affects neuronal development.
The clinical manifestations of WMI are spastic diplegia or tetraplegia due to damage of corticospinal tract axons, visual impairment due to damage of the optic radiations, cognitive deficits, and seizures. Their severity correlates with pathology and is most pronounced in cystic white matter necrosis. These deficits may not be apparent initially. They may be fully appreciated months or years after the injury occurs. WMI is the substrate of cerebral palsy. The leading risk factor in 75% of CP cases is prematurity and the underlying pathology in most of CP is WMI.
Our concept of WMI has evolved over the past 50 years. Initially, it was conceived of as an acute ischemic lesion with cavitated white matter infarcts. Now, it is a spectrum of pathology some of which is caused by chronic hypoxia-ischemia or infection and results in white matter damage and neuronal loss.
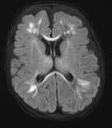 HPeV-3 meningoencephalitis |
Necrotic white matter lesions similar to those caused by hypoxia-ischemia are caused by the Picornavirus Human Parechovirus 3 (HPeV-3) which is the most common cause of viral meningoencephalitis in young infants. HPeV-3 causes cyclic outbreaks and nosocomial infections every 2-3 years, characterized by high fever, irritability, rash and seizures. There is no CSF pleiocytosis, and PCR is required for diagnosis. White matter necrosis is due to the direct effects of the virus.
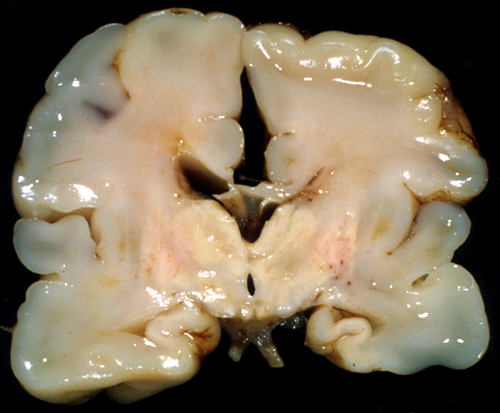 HIE-thalamic and hippocampal lesions |
A distinction is often made between HIE in premature infants, which causes mainly WMI, and HIE in mature infants, which affects predominantly the cortex and deep nuclei. This is an oversimplification. Pathological and volumetric MRI studies show that, in WMI, there is loss of neurons (and reduction in mass) in the cortex and deep nuclei, especially the thalamus. Moreover, as the picture on the left illustrates, the full range of gray matter HIE pathology that occurs at term may also develop in premature infants, usually along with white matter damage.
Further Reading
- Banker BQ, Larroche J-C. Periventricular leukomalacia
of infancy. A form of neonatal anoxic encephalopathy.
Arch Neurol 1962;7:386-10.
Woodward LJ, Andeson PJ, Austin NC et al. Neonatal MRI to predict neurodevelopmental outcomes in preterm infants. N Engl J Med 2006;355:685-94. PubMed - Leviton a, Gressens P. Neuronal damage accompanies white matter damage. Trends in Neurosciences 2007;30:473-8. PubMed
- Khwaja O, Volpe, JJ. Pathogenesis of cerebral white matter injury of prematurity. Arch Dis Child Fetal Neonatal Ed 2008;93:F153-6. PubMed
- Volpe JJ. Encephalopathy of Congenital Heart Disease– Destructive and Developmental Effects Intertwined. J Pediatr 2014; 164: 962-965.PubMed
- Morton PD,Nobuyuki I,Jonas RA, Gallo V. Congenital cardiac anomalies and white matter injury. Trends in Neurosciences 2015;38 (6):353-363. PubMed
- Ortinau C, Neil J. The Neuroanatomy of Prematurity:Normal Brain Development and the Impact of Preterm Birth. Clinical Anatomy 2015; 28:168-183.PubMed
- Volpe JJ, Kinney HC, Jensen FE, Rosenberg PA. The developing oligodendrocyte: key cellular target in brain injury in the premature infant. Int J Dev Neurosci. 2011; 29(4): 423–440. PubMed
- Back SA. White Matter Injury in the Preterm Infant: Pathology and Mechanisms. Acta Neuropathol. 2017; 134(3): 331–349.PubMed
- Inder TE, Volpe JJ, and Anderson PJ. Defining the Neurologic Consequences of Preterm Birth. N Engl J Med.2023 Aug 3;389(5):441-453. PubMed
Updated: August, 2023