ASPHYXIA AND HYPOXIC-ISCHEMIC ENCEPHALOPATHY IN MATURE INFANTS
In adults, the brain is about 2% of body weight and receives about 15% of the cardiac output. In term babies, it is 10% of body weight and uses energy not only to maintain electrical activity but also for growth.
The pathogenesis of HIE in the newborn period involves the same key players that are also present in adults, namely energy crisis from hypoxia-ischemia, lactic acidosis, excitotoxicity, and free radicals, but there are some important modifiers, which account for different patterns of pathology in newborn babies. Antioxidant defense mechanisms are not fully developed in newborns; the patterns of glutamatergic neurotransmission are evolving and glutamate receptors are more abundant in deep nuclei than in the cortex; myelin is largely absent in the centrum semiovale; the newborn brain has a much higher water content.
In many mature infants, HIE is caused by events that occur during labor and delivery. Pregnancies advance uneventfully to term and then, for no apparent reason, after a flurry of unusually vigorous activity, fetal movement decreases or ceases. The fetus dies in utero, or is delivered aphyxiated, usually after emergency Cesarean section.
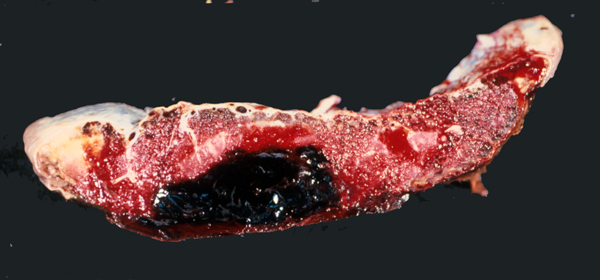 HIE. Placental abruption |
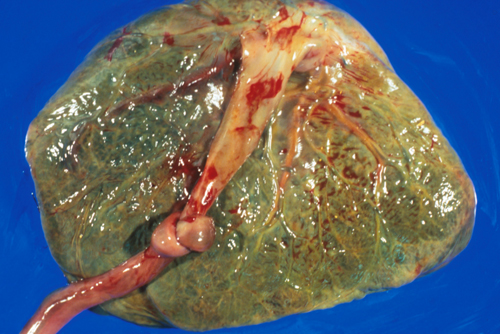 HIE. Umbilical cord knot and meconium staining |
In some cases, the cause of HIE or fetal loss is obvious, e.g., placental abruption, cord accident, or presumed cardiovascular stress during a difficult delivery. In many others, it is difficult to pinpoint. Prenatal factors (eclampsia, hypercoagulability and other maternal conditions, placental pathology) increase the risk of an adverse outcome, but neonatal encephalopathy and fetal death can occur without such predisposing factors. In a few cases, neonatal encephalopathy develops in the postnatal period as a result of septic shock, persistent fetal circulation, and other causes. Also, pathology that develops initially before or during birth, e.g., meconium aspiration, plays out in the postnatal period, adding to the initial insult.
 HIE. Borderzone lesions |
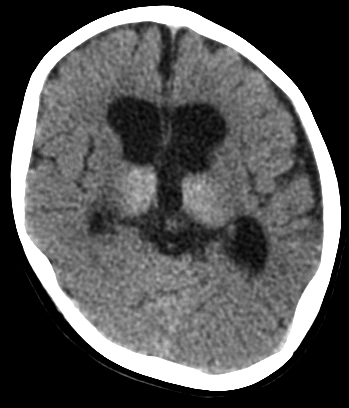 HIE. Thalamic involvement |
 HIE in MCA territory |
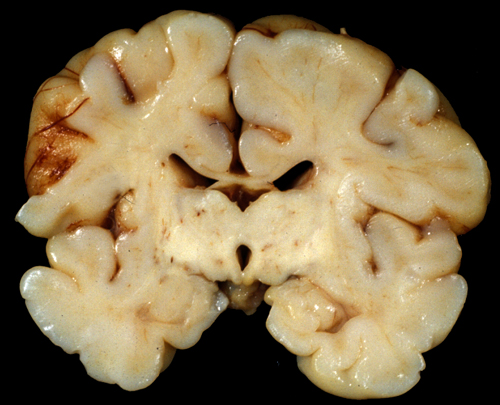
Two main patterns of brain damage have been defined by MRI and neuropathological studies: moderate ischemia causes mainly cortical damage in the border zones between major arterial territories (a parasagittal band of cortex that arches from the frontal to the occipital pole); severe ischemia damages the deep nuclei and the brainstem. These patterns correspond to the neuropathology seen in animal models of perinatal asphyxia. While lesions that confined to these patterns are seen in many cases, frequntly, the topography of damage varies and the two patterns overlap: cortical lesions are more extensive and irregular than the parasagittal area or they fall within rather than between vascular territories, and damage of deep nuclei is accompanied by cortical injury. However, unlike adults, the basal ganglia, thalamus and brainstem of mature infants are affected more severely than the cerebral cortex and hippocampus, probably because they have higher energy demands and more developed neurotransmission compared to the cortex. A similar predilection for the basal ganglia and brainstem is seen in the Leigh Syndrome, a mitochondrial encephalopathy that affects young children. Also, similar patterns of injury are seen sometimes in premature infants along with white matter pathology.
In adults, HIE causes cortical atrophy, but the basic structure of the brain and the gyral pattern are preserved.
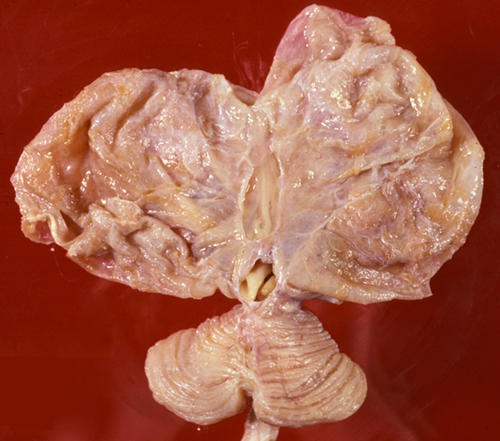 Severe brain atrophy from perinatal HIE |
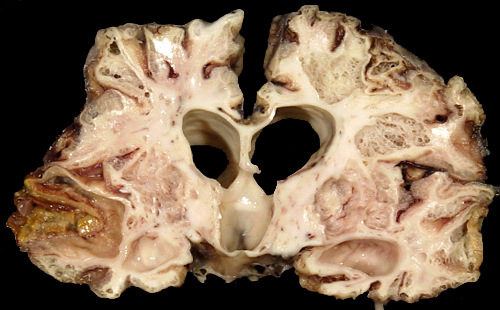 Multicystic encephalopathy. |
 Multicystic encephalopathy. |
In mature infants, severe perinatal HIE can cause the cortex and white matter to melt away to such an extent that the brain is reduced to a thin-walled sac, similar to hydranencephaly. In other cases, destruction of cortex and white matter results in the formation of multiple cavities traversed by a web of delicate glial strands. The cavities contain fluid, debris, and macrophages. This entity is called cystic encephalomalacia or multicystic encephalopathy.
 Ulegyria |
 Ulegyria-mushroom gyri |
 Status marmoratus |
Cortical damage is always more severe in the deeper parts of sulci while the crowns of gyri are less affected. This leads to formation of mushroom-shaped gyri. The visible crowns of these gyri may be relatively normal or atrophic but their deeper parts are undermined. The term "ulegyria" (scarred gyri) refers to gyral atrophy and gliosis. Thalamic and basal ganglia damage causes, over time, loss of neurons, mineralization of damaged neurons, and gliosis. An abnormal or excessive pattern of myelin develops in some cases. Irregular patches of dense myelin mixed with gliotic zones give the thalamus and basal ganglia a marbled appearance to the naked eye, a condition called status marmoratus. The hippocampus is less frequently affected than in adult HIE. In a few cases, however, combined damage of the hippocampus and pons (pontosubicular necrosis) is seen. The pathogenesis of this unusual pattern is unclear. In some cases, perinatal HIE causes bilateral loss of hippocampal pyramidal neurons, similar to adult HIE. The MRI in such cases shows hippocampal atrophy. This lesion causes developmental amnesia.
DEVELOPMENTAL AMNESIA (DA):
A memory disorder caused by bilateral
hippocampal damage resulting from
HIE in the perinatal period or later
in childhood. DA can occur without
cerebral palsy. Some patients have
seizures. A minimum of 25-30% loss
of hippocampal volume is required
to cause DA.
DA impairs episodic memory
(remembering events of everyday life).
Semantic memory (memory for facts)
is relatively preserved. Visual and
verbal recognition is preserved; visual
and verbal recall is impaired. Immediate
memory is intact. DA is milder when
HIE occurs early and more severe when
HIE occurs later in childhood, probably
because of higher plasticity with
early lesions.
DA may be missed in the first few
years of life and only become noticed
upon entering school. Despite their
disability, DA patients may be able
to retain factual information and
acquire language skills. Their problems
may be attributed to absentmindedness,
and specialized neuropsychological
tests are needed to reveal the memory
impairment.
The outcome of neonatal encephalopathy correlates with the topography of the lesions. Brainstem injury usually causes death in the newborn period, because of damage of vital centers of respiration and cardiac function. Infants with cortical lesions survive but have intellectual disability and cerebral palsy. Given the central role of the thalamus in cognition and consciousness, severe injury leads into a persistent vegetative state.
The timing of injury is an important issue, especially in law suits involving neonatal encephalopathy. There is limited literature, based on animal experiments and human observations, that allows a rough timing of the pathology while it is still evolving in the first two weeks. After this, it is difficult to distinguish between months or years. These determinations are now based mostly on the MRI findings.
Further Reading
- Ferriero DM. Neonatal brain injury. N Engl J Med 2004;351:1985-95. PubMed
- Cowan F, Rutherford M, Groenedaal F, et al. Origin and timing of brain lesions in term infants with neonatal encephalopathy. Lancet 2003;361736-42. PubMed
- Ellis WG, Goetzman BW, Lindenberg JA. Neuropathological documentation of prenatal brain damage. AJDC 1988;142:858-66. PubMed
- Gadian DG, Aicardi J, Watkins K et al. Developmental amnesia associated with early hypoxic-ischemic injury. Brain 2000;123:499-507. PubMed
- Volpe JJ. Neonatal Encephalopathy: An Inadequate Term for Hypoxic�Ischemic Encephalopathy. Ann Neurol 2012;72:156�166. PubMed
Updated: January, 2013