CEREBRAL INFARCTS
Cerebral infarction is focal brain necrosis due to complete and prolonged ischemia that affects all tissue elements, neurons, glia, and vessels.
CLINICAL FINDINGS
Ischemic infarcts cause focal neurological deficits. In embolic infarcts, these appear abruptly. In atherothrombotic infarcts, they evolve over a period of time, usually over hours. Atherothrombotic infarcts are often preceded by transient ischemic attacks (TIAs). A TIA is a focal neurological deficit that lasts less than 24 hours and resolves. The mechanism of TIAs is uncertain. They may be caused by critical reduction of perfusion that impairs neurological function but falls short of causing permanent tissue damage, or by emboli that break up soon after they occlude vessels.
The release of osmotically active substances (arachidonic acid, electrolytes, lactic acid) from the necrotic brain tissue causes cerebral edema. This is aggravated by vascular injury and leakage of proteins in the interstitial space. By 3-4 days, interstitial fluid accumulates in the infarct and around it. This is the most dangerous period for a large cerebral infarct. Death from a massive hemispheric infarct is caused by cerebral edema and herniations, not by the loss of brain tissue. Recovery of function, after an infarct, is due initially to restoration of perfusion in the penumbra (see below) and then to settling down of cerebral edema. Additional improvement may occur later through mechanisms involving neuronal plasticity.
The basic mechanisms of cell and tissue injury that were discussed under HIE apply also to infarcts. One additional concept, the ischemic penumbra, is worth stressing. In every infarct, there is a central core of total ischemia and necrosis which is irreversible. This area is surrounded by a zone of borderline ischemic tissue, the ischemic penumbra. Ischemia, in the penumbra, causes dysfunction due to ionic and metabolic disturbance but is not severe enough to result in structural damage. Prompt restoration of perfusion in the penumbra by injection of thrombolytic agents may prevent structural damage in this area, thus limiting the neurological deficit. Ischemic stroke is an emergency. The window of opportunity for salvaging the penumbra is very short. If adequate blood supply is not restored within 3 hours, necrosis extends to the penumbra.
PATHOLOGY OF ISCHEMIC INFARCTS
In the first day or so, the infarct appears as a poorly demarcated area of softening.
 Acute Right MCA infarct |
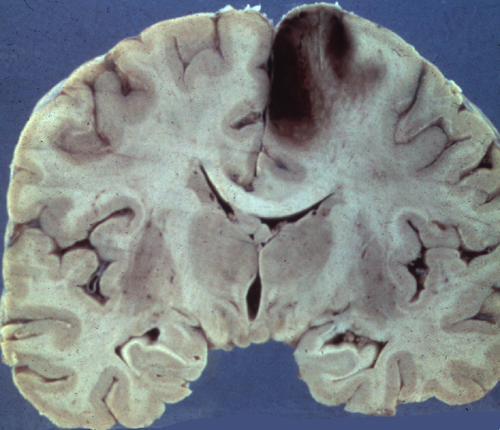 Acute RACA infarct |
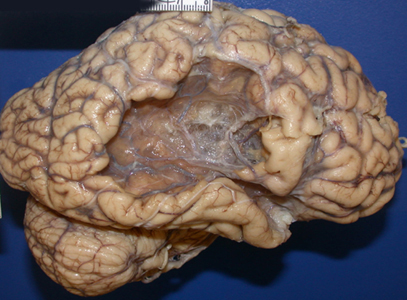 Old right MCA infarct |
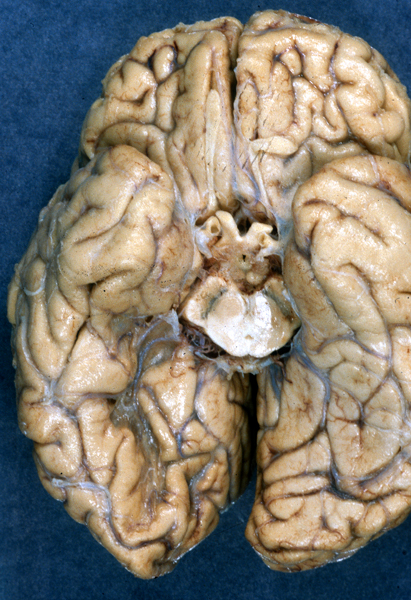 Old right PCA infarct |
CT Imaging at this stage may be negative, especially in brain stem infarcts. MRI is much more sensitive. At the peak of edema, the infarct appears hypodense and bright on T2 MRI images. The infarcted tissue becomes sharply demarcated and softens progressively. From the second week onward, it begins to disintegrate and is gradually replaced by a cavity. The size and location of infarcts follows the anatomy of vascular territories. The territories of the three main cerebral arteries are illustrated in the pictures to the left. Occlusion of vessels supplying the brainstem and cerebellum causes infarcts with characteristic patterns and clinical deficits that correspond to the nuclei and tracts that are affected (see supplemental material).
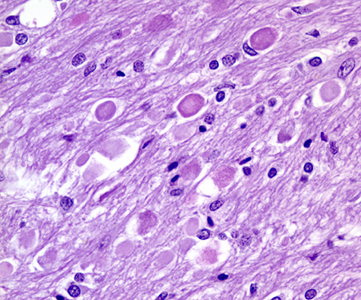 Axonal swellings |
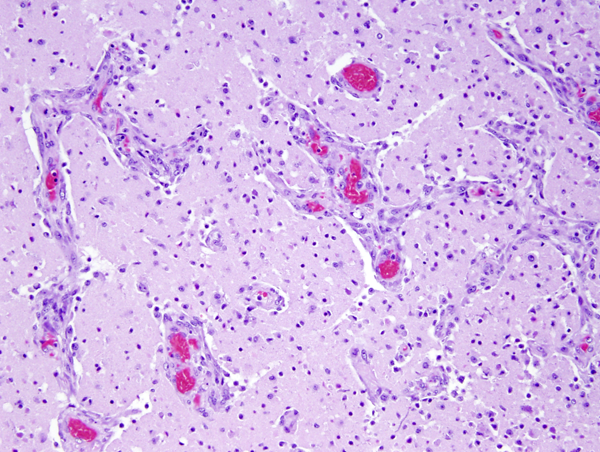 Neovascularization |
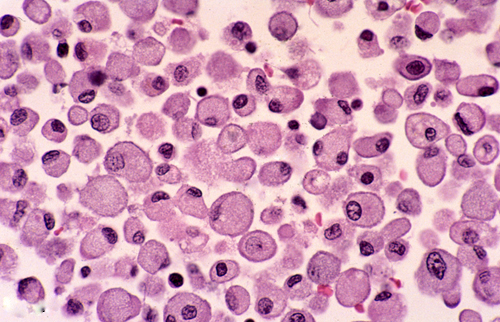 Macrophages |
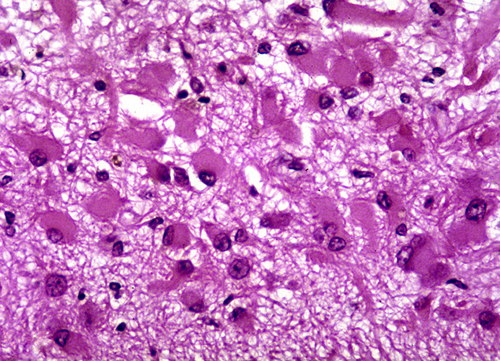 Gemistocytic astrocytes |
Microscopical examination in the first 24 to 48 hours reveals anoxic neurons, pallor of staining and vacuolization of the white matter due to unraveling of myelin, and axonal swellings. During the first week, there is a transient inflammatory reaction, especially around blood vessels and in the meninges, due to release of arachidonic and other fatty acids. As the core of the infarct disintegrates, endothelial cells from the periphery proliferate, and capillaries grow into the dead tissue. Neovascularization (which accounts for contrast enhancement) peaks at 2 weeks. Monocytes from the blood stream enter the infarct through damaged vessels. They ingest the products of degradation of neurons and myelin and are transformed into lipid-laden macrophages. Macrophage reaction appears early and peaks at 3-4 weeks. Astrocytes from the surrounding undamaged brain proliferate and form a glial scar around the infarct. This is completed in approximately 2 months. After that, the infarct remains unchanged. With maturation of new capillaries and glial scar formation, the blood-brain barrier is once again sealed. Neurons do not regenerate. So, some brain tissue is lost forever.
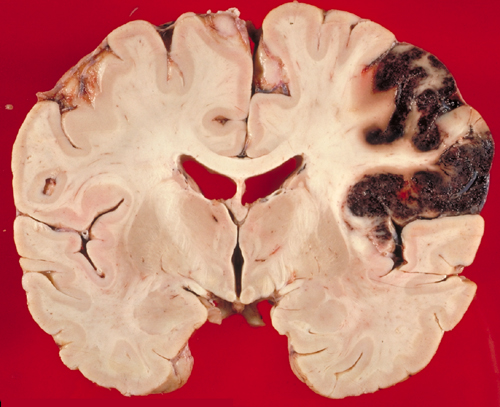 Hemorrhagic infarct |
A hemorrhagic infarct is an infarct stippled with petechiae or showing confluent larger hemorrhages, especially in necrotic gray matter. Blood leaks from collateral vessels or through necrotic capillaries when the occluding thrombus or embolus breaks up and the infarcted area is reperfused. Hemorrhagic infarcts are most common in embolism. Use of thrombolytics or anticoagulants may convert a bland infarct into a hemorrhagic one.
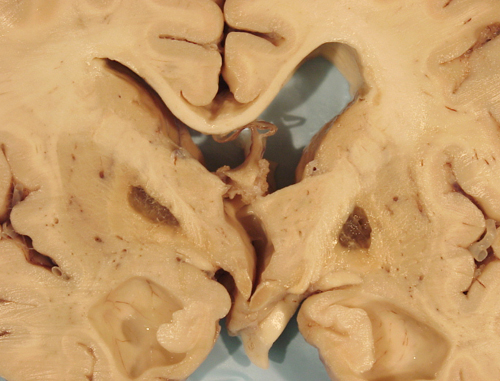 Lacunar infarcts |
Lacunar infarcts are small infarcts in the deeper parts of the brain (basal ganglia, thalamus, white matter) and in the brain stem. They are responsible for about 20 percent of all strokes. They are caused by occlusion of deep penetrating branches of major cerebral arteries and are particularly common in hypertension and diabetes, which are associated with severe atherosclerosis of small vessels and small vessel disease (see below). A small lacunar infarct (e.g., one involving the internal capsule) can cause as severe a neurological deficit as can a much larger hemispheric infarct but without the life-threatening cerebral edema that is seen in the latter.
CAUSES OF ISCHEMIC INFARCTS
The diverse types of vascular disease and other conditions that cause cerebral infarction are partially listed in the table below and briefly discussed further on. Clinical classification systems for ischemic stroke, such as the TOAST (Trial of Org 10170 in Acute Stroke Treatment) and the ASCO (Atherosclerosis, Small vessel disease, Cardiac source, Other cause) systems have been introduced. These systems are based on clinical and ancillary findings, including noninvasive angiography and transthoracic echocardiography. Atherothrombosis, small vessel disease, and cardioembolism are the most common causes of ischemic stroke in elderly patients. Cardioembolism and the other entities listed can cause stroke in younger individuals, and some of them even in children.
| Atherosclerosis-atherothrombosis |
| Small vessel disease |
| Cardioembolism, including paradoxical embolism through a patent foramen ovale |
| Vasculitis |
| Arterial dissection |
| Polycythemia, thrombocythemia, TTP |
| Systemic lupus erythematosus |
| Sickle cell disease |
| Vascular spasm |
| Meningitis and fungal vasculitis |
| Hypercoagulability (Factor V Leiden, Prothrombin 20210A mutation, antiphospholipid antibody syndrome) |
| Inherited metabolic disorders (Fabry disease, homocystinuria, mitochondrial disorders) |
| Fibromuscular dysplasia and other angiopathies |
 Severe atherosclerosis |
 Atherosclerosis and thrombosis |
A large proportion of infarcts are caused by atherosclerosis of large arteries, alone or with superimposed thrombosis. Atherosclerosis involves the circle of Willis and large leptomeningeal arteries and extends into their smaller branches. Atheromatous plaques begin with intimal thickening and accumulation of blood-derived lipids in the intima, followed by disruption of the interna elastica, accumulation of lipid laden macrophages, formation of cholesterol crystals, and deposition of calcium. The process of lipid accumulation is accompanied by an inflammatory reaction involving lymphocytes and macrophages. Atheromatous plaques may cause narrowing or occlusion of the vascular lumen by themselves or after rupture and thrombosis. Cholesterol crystals from ruptured plaques may embolize distal vessels.
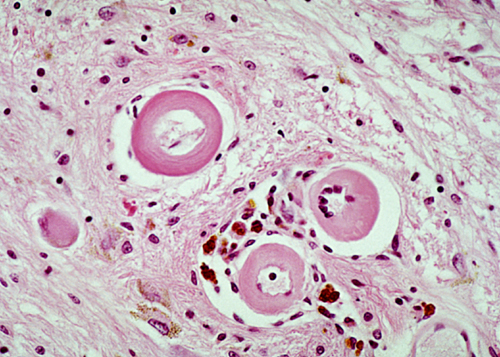 Arteriolosclerosis |
Small vessel disease includes atherosclerosis of small arteries but refers more specifically to arteriolosclerosis, a vascular lesion that is seen primarily in hypertension and diabetes but also occur in old age without these predisposing conditions. This pathology affects small penetrating arteries and arterioles that originate from the base of the brain and supply the basal ganglia, thalamus, deep white matter, and the brainstem. Affected vessels become thickened and stiff, and the normal components of their walls are replaced by a homogeneous, glassy (hyaline) substance, composed of collagen and other proteins. The pathogenesis of this change varies: in hypertension, it is caused by endothelial injury and leakage of plasma proteins in and around vessels; in diabetes, it probably has to do with glycation of proteins and diffuse basement membrane thickening. Its effects are narrowing of the lumen and tortuosity, which lengthens the distance blood has to travel to perfuse its targets. Ischemia, resulting from these processes, causes small infarcts (lacunar infarcts) and diffuse loss of axons and myelin, causing vacuolation of the white matter (leukoaraiosis, thinning out of the white matter). Loss of contractility from the destruction of pericytes and smooth muscle impairs regulation of blood flow. Loss of elasticity leads to development of small aneurysms and makes vessels fragile, resulting in microbleeds and large catastrophic hemorrhages, which occur spontaneously or after trivial trauma. The MRI findings in arteriolosclerosis are periventricular and subcortical white matter hyperintensities on FLAIR imaging, lacunes, enlarged perivascular spaces and cerebral microbleeds. The brain damage from arteriolosclerosis causes cognitive impairment. Genetic angiopathies further on. See also the white matter in HIE.
According to some authors, embolism is the most frequent cause of ischemic infarction. Most emboli are fragments of blood clots that originate in the heart or major vessels. Conditions causing cardiac emboli include myocardial infarcts, atrial fibrillation and other arrhythmias, rheumatic heart disease, bacterial and non-bacterial endocarditis, prosthetic valves, mitral valve prolapse, atrial myxoma, calcified mitral annulus, and cardiomyopathy. An embolus cannot be distinguished grossly or microscopically from a locally formed thrombus. An infarct is assumed to be embolic if it is hemorrhagic, there is a source of emboli, there are multiple infarcts of the brain and other organs (kidney, spleen), and there is no atherosclerosis or other vascular disease. Some emboli consist of atheromatous material that is detached from ulcerated atheromas of the aorta or carotid arteries. Vascular manipulation (angiography, carotid endarterectomy) may cause atheromatous embolism. Rarer causes of embolism are fat, air, and tumor emboli. Unlike atherothrombotic infarcts, which may evolve within hours or days, embolic infarcts have an abrupt onset.
Vasculitis
CNS vasculitis can be classified under the following categories:
 Temporal (giant cell) arteritis |
 Aspergillus arteritis |
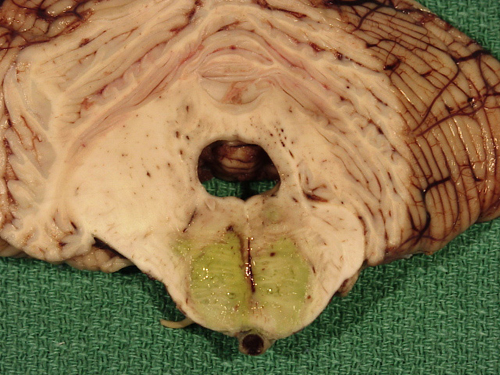 Mucor arteritis of the basilar artery and pontine infarct |
A. Systemic vasculitis with CNS involvement: giant cell arteritis, polyarteritis nodosa, Wegener granulomatosis, Takayasu arteritis, Kawasaki disease. The most common of these entities is giant cell (temporal) arteritis (GCA) which is more frequent in older people and women and is associated with polymyalgia rheumatica. GCA is also called temporal arteritis because it frequently involves the temporal artery. Biopsy of the temporal artery is done for diagnosis. However, GCA affects the aorta and other major extracranial and, less frequently, intracranial branches. Involvement of the ophthalmic artery causes visual loss in a significant number of cases. GCA is a T-cell mediated autoimmune condition that affects medium-size and large arteries. Lymphocytes and multinucleated giant cells infiltrate the vessel wall, disrupt the internal elastic lamina, and cause narrowing and thrombosis.
B. Secondary vasculitis: Infectious (bacterial, fungal, spirochetal, viral); collagen-vascular disease (SLE, rheumatoid arthritis, Behcet disease); drug-induced vasculitis. Fungal vasculitis is most frequently caused by aspergillus and mucor.
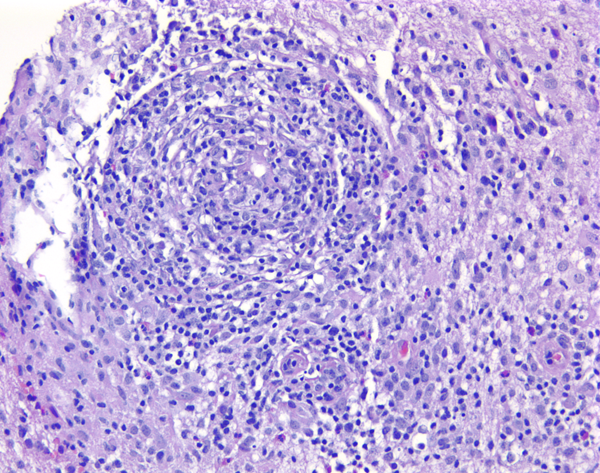 Cerebral vasculitis |
C. Primary angiitis of the CNS (PACNS). PACNS presents clinically with focal neurologic deficits, headaches, encephalopathy, and seizures. Angiographic or pathological documentation of vasculitis, and absence of evidence of systemic or secondary vasculitis are criteria for diagnosis. Some cases of PACNS involve medium-to-large cerebral vessels and can be diagnosed by angiography and MRI imaging showing vascular stenoses and ischemic or hemorrhagic strokes. Other cases of PACNS affect primarily small cortical and leptomeningeal blood vessels. Small vessel PACNS presents with focal or diffuse neurologic abnormalities and enhancing meningeal and parenchymal lesions. The clinical picture overlaps encephalitis. Angiography is normal in such cases and brain biopsy is required for diagnosis. By definition, all cases of angiitis have intramural, usually mononuclear, inflammation. There is usually also perivascular inflammation that may be difficult to distinguish from perivascular mononuclear infiltrates that are seen in MS and infectious diseases. Some cases of small vessel PACNS show also epithelioid cell granulomas with multinucleated giant cells and amyloid deposits in vessel walls (granulomatous PACNS). Paraneoplastic vasculitis, similar to PACNS, may occur rarely in association with lung, gastrointestinal and other cancers.
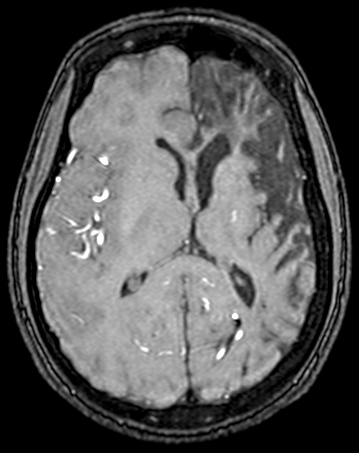 MCA-ACA infarct in sickle cell disease |
Other causes of arterial occlusion and infarction include:
Hematologic disorders - Polycythemia, hemoglobinopathies (sickle cell disease), deficiencies of anticoagulant factors, thrombotic thrombocytopenic purpura.
Metabolic disorders - Dyslipoproteinemias, Fabry disease, homocystinuria, organic acidemias, mitochondrial disorders. Some of these conditions cause ischemic infarcts even in children and infants. Mitochondrial disorders can cause TIAs and ischemic strokes.
Hereditary hypercoagulability disorders: Factor V Leiden, Prothrombin 20210A. These polymorphisms derange the delicate balance between natural anticoagulant and procoagulant pathways. They are very prevalent in the population and combine with one another and with acquired conditions that promote clotting, causing venous and arterial infarcts.
 Dissecting aneurysm |
Trauma to the head and neck can cause dissecting aneurysms and other lesions of the carotid and vertebral arteries. The pattern of brain necrosis in severe traumatic brain injury often suggests vascular occlusion. In some cases, arterial dissection occurs apparently spontaneously, without a traumatic event.
Contraceptives and estrogen therapy cause most commonly venous thrombosis and rarely intimal hyperplasia and thrombosis of cerebral and extracerebral arteries.
Vascular Spasm. This is a complication of subarachnoid hemorrhage.
Genetic angiopathies : Cerebral autosomal dominant arteriopathy with subcortical infarcts and ischemic leukoencephalopathy (CADASIL), caused by mutations of the NOTCH3 gene, is a small vessel arteriopathy in which deposition of a granular osmiophilic material in the vessel wall causes loss of smooth muscle, thickening of the wall, and narrowing of the lumen. The symptoms are due to involvement of the brain but the pathology affects other organs and tissues and can be diagnosed with a skin biopsy. An autosomal dominant angiopathy due to mutations of COL4A1, which encodes a collagen of the vascular adventitia, causes porencephaly in infants and a spectrum of disease in adults that includes small vessel disease, lacunar infarcts, microhemorrhages, deep intracerebral hemorrhages, intracranial aneurysms, and leukoencephalopathy. Other COL4A1 related disorders are renal and liver cysts, and eye abnormalities. Cerebral amyloid angiopathy, caused by deposition of various types of amyloid, causes similar vascular pathology but affects primarily leptomeningeal and cortical vessels.
Miscellaneous: Spontaneous dissecting aneurysms, moyamoya disease (narrowing of proximal cerebral arteries).
The fat embolism syndrome (FES) is a complication of long bone fractures, orthopedic surgery, and traumatic lesions of the viscera and subcutaneous tissue, including burns. The most common non-traumatic cause of the FES in children and young individuals is sickle cell disease. Other non-traumatic settings of FES include, osteomyelitis, pancreatitis, and diabetes. Clinically, FES is characterized by petechial rash, hypoxemia, deterioration of mental status, and thrombocytopenia. Pathologically, there are petechial hemorrhages and micro-infarcts in the brain, heart, and other organs.
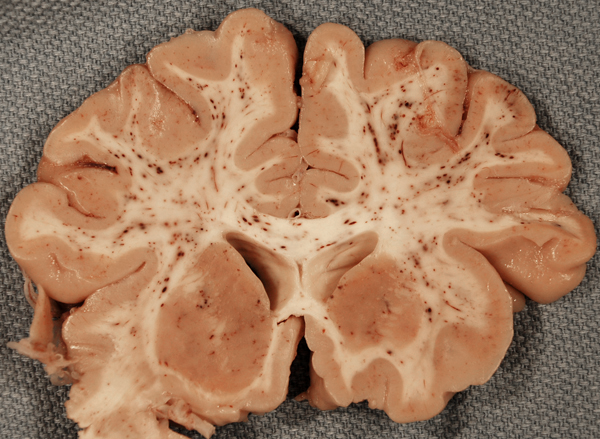 Fat embolism syndrome |
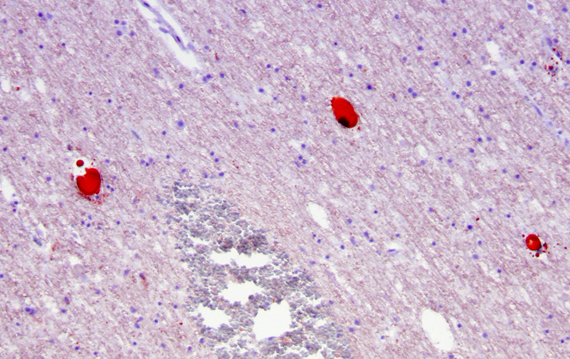 Fat embolism syndrome |
Fat, mobilized during trauma or osteonecrosis, enters the venous circulation and embolizes pulmonary artery branches and capillaries. If the capacity of pulmonary capillaries is exceeded or the lungs are by-passed through a patent foramen ovale, fat globules enter the arterial circulation and occlude capillaries in the heart, brain, kidneys, and other organs, causing hemorrhagic and ischemic lesions. There is some debate about the pathogenesis of the FES. Some attribute it to mechanical obstruction by fat globules. Other views maintain that it is caused by agglutination of chylomicrons and toxic action of free fatty acids and other fat breakdown products.
CEREBRAL VENOUS THROMBOSIS
 Sinovenous thrombosis |
Venous thrombosis involves more commonly the venous sinuses and less frequently their tributary cortical veins and causes congestion, hemorrhage, and necrosis of brain tissue that does not correspond to vascular territories (venous infarction) and increased intracranial pressure. Thrombosis of the superior sagittal sinus causes parasagittal infarcts resulting in paresis of one or both legs. The causes of venous thrombosis are diverse and include inherited thrombophilias, the antiphospholipid syndrome, pregnancy and the postpartum state, oral contraceptives and cancer. In infants and children, venous thrombosis is most commonly caused by dehydration from diarrhea. Sinovenous thrombosis accounts for some cases of the syndrome of pseudotumor cerebri, which is characterized by headache, papilledema, increased CSF pressure and normal size ventricles. These symptoms and signs are caused by intracranial hypertension which is due to impaired resorption of CSF into the venous sinuses and venous congestion. This syndrome has many other causes, including meningeal pathology, tumors, toxic and metabolic disorders, and conditions that cause marked elevation of CSF protein, such as the Guillain-Barré syndrome. In many cases, no specific etiology is identified (idiopathic intracranial hypertension). Rare cases of cerebral venous thrombosis have been reported in patients receiving adenoviral vector vaccines against Covid-19. These cases are associated with thrombocytopenia and are mediated by antibodies to platelet factor 4.
VASCULAR DEMENTIA
Vascular dementia is the second most common cause of dementia after Alzheimer’s disease. It may be present alone but most commonly coexists with neurodegenerative diseases such as Alzheimer’s disease, Parkinson’s disease and frontotemporal dementia. Indeed, most cases of dementia in old persons are multifactorial and in these cases the vascular pathology is thought to account for about one third of the total. It is due to loss of brain tissue from large infarcts, multiple small infarcts (multi-infarct dementia), microinfarcts, and leukoencephalopathy. These lesions affect cumulatively large areas of the brain, especially regions involved in memory and higher functions. The underlying vascular pathology is atheroembolic disease, arteriolosclerosis, cerebral amyloid angiopathy and other vasculopathies. Cortical atrophy and hippocampal sclerosis may also be present. The main predisposing factors are old age and hypertension. See also "Small Vessel Disease" and "the White Matter in HIE" in this chapter.
Further Reading
- Ringelstein EB, Nabavi DG. Cerebral small vessel diseases: cerebral microangiopathies. Curr Opin Neurol. 2005;18:179-88. PubMed
- Adams HP Jr, Bendixen BH, Kappelle LJ, et al. Classification of subtype of acute ischemic stroke: definitions for use in a multicenter clinical trial: TOAST: Trial of Org 10172 in Acute Stroke Treatment. Stroke 1993;24:35– 41.PubMed
- Amarenco P, Bogousslavsky J, Caplan LR, et al. New approach to Stroke Subtyping: The A-S-C-O (Phenotypic) Classification of Stroke. Cerebrovasc Dis 2009;27:502-8. PubMed
- Chabriat H, Joutel A, Dichgans M, et al. CADASIL. Lancet Neurol 2009;8:643�53. PubMed
- Guida A, Tufano A, Perna P, et al. The thromboembolic risk in giant cell arteritis: a critical review of the literature. Int J Rheumatol. 2014; 2014: 806402. PubMed
- Jaunmuktane Z, Mahadeva U, Green A, et al. Microvascular injury and hypoxic damage: emerging neuropathological signature of COVID-19. Acta Neuropathologica 2020;140:397-400.PubMed
- Blevins BL, Vinters HV, Love S et al. Brain arteriolosclerosis. Acta Neuropathologica 2021; 141:1–24PubMed
Updated: March, 2023