SPECIFIC NEUROPATHIES
DIABETIC NEUROPATHIES
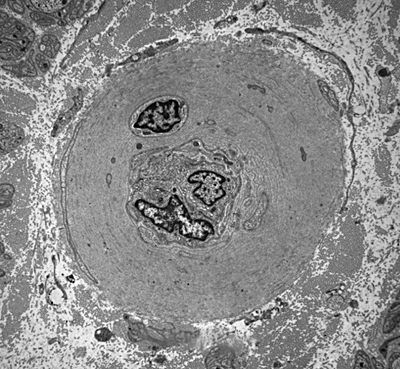 Arteriole in diabetic nerve |
The most common cause of neuropathy in clinical practice is diabetes. Peripheral neuropathy develops in more than half of long term diabetics and goes in tandem with other complications, such as retinopathy and nephropathy. Diabetes causes several types of neuropathy, which include chronic symmetrical polyneuropathy, proximal neuropathy (diabetic amyotrophy), mononeuropathies, and cranial radiculopathies. The most common is a symmetrical sensorimotor neuropathy which causes pain, sensory loss, weak or absent tendon reflexes and, to a lesser extent, weakness. The pathological findings in this diabetic polyneuropathy are axonal loss, axonal regeneration, and in some patients, demyelination. Small axons are affected more severely.A prominent finding in diabetic neuropathy is thickening of endoneurial arterioles due to increased deposition of basement membrane material, similar to changes that occur in brain arterioles and glomerular capillaries. The pathogenesis of diabetic neuropathies is poorly understood. Ischemia is probably an important factor. Nonenzymatic glycation of neural structures and other biochemical changes in diabetes probably play a role also.
INFLAMMATORY DEMYELINATIVE NEUROPATHIES
These uncommon neuropathies are presumed to be immune disorders in which antibodies and activated T-lymphocytes, reacting with antigens present on peripheral nerves, elicit an inflammatory and macrophage reaction that destroys myelin and axons. The strongest evidence of a humoral immune reaction in these neuropathies is that plasma exchange results in significant clinical improvement. The participation of cellular immunity is underlined by the pesence of T-lymphocytes around blood vessels in affected nerves. The two main entities in this group are the Guillain-Barré syndrome and chronic inflammatory demyelinative neuropathy. An experimental model of demyelinative neuropathy, experimental allergic neuritis (EAN), can be produced by injecting animals with myelin and Freund adjuvant or purified peripheral myelin protein P2. EAN is a cell-mediated immune reaction.
 Guillain-Barré syndrome |
The Guillain-Barré Syndrome(GBS) is not a single disease entity. It includes Acute Inflammatory Demyelinative Polyneuropathy (AIDP), Acute Motor Axonal Neuropathy (AMAN), the Miller-Fisher syndrome-MFS(acute ophthalmoplegia, areflexia and ataxia)and several other variants. AIDP accounts for 90% of GBS. It is rare but represents a medical emergency in which prompt diagnosis may save lives. It begins with paresthesias in the toes and fingertips, followed by rapidly advancing weakness and areflexia. Weakness reaches a plateau within four weeks, after which recovery begins. Some cases are fulminant, evolving in one or two days. At the height of their disease, many patients are completely paralyzed and unable to breathe. Even with modern intensive care, approximately 5% of patients die from respiratory paralysis, cardiac arrest (probably due to autonomic dysfunction), sepsis, and other complications. Ten percent of those who recover have residual weakness. Though easy to diagnose in its classical form, GBS is often missed because of atypical clinical features which include ophthalmoplegia, ataxia, sensory loss, and dysautonomia. Plasma exchange (presumably removing the offending antibodies) and intravenous immunoglobulin are the treatments of choice. The two key laboratory abnormalities in GBS are decreased nerve conduction velocity or conduction block and elevated CSF protein with relatively few cells (albuminocytologic dissociation).
Peripheral nerves show perivenular mononuclear cells, demyelination (myelin proteins are the source of elevated CSF protein), and macrophages. Axonal damage, which accounts for the permanent deficits, is variable and may be severe. The pathology is most severe in spinal roots and plexuses and less pronounced in more distal nerves. In the phase of recovery, the nerve contains thin myelin sheaths, indicating myelin regeneration. AMAN shows axonal damage with little inflammation.
Most cases of GBS are triggered by a previous bacterial or viral infection and the most common trigger, accounting for about 30% of cases is Campylobacter Jejuni (CJ), the leading cause of bacterial gastroenteritis in developed counties. Less frequently, GBS is triggered by Mycoplasma pneumoniae, Cytomegalovirus (CMV) and other viral infections or vaccinations. The oligosaccharides that coat the bacterial wall of CJ are similar to GM1 ganglioside that is present in the axonal membrane at the nodes of Ranvier and in paranodal myelin. So antibodies against CJ generated in the course of the infection cross-react with GM1 ganglioside in peripheral nerves and elicit inflammation that damages these structures. Anti-GM1 antibodies are also found in the serum of GBS patients. GBS following CMV infections has anti-GM2 antibodies.
Chronic inflammatory demyelinative polyradiculoneuropathy (CIDP) follows a chronic or relapsing course over many months or years and may cause severe permanent disability. Nerve conduction studies show decreased conduction velocity, conduction block, and prolonged distal latencies and F waves. In the active phase of the disease, the CSF shows elevated protein without increased cells. Pathologically, peripheral nerves show demyelination, thin (incompletely regenerated) myelin, and hypertrophic changes due to recurrent attacks of demyelination with intervals of repair. In chronic cases, there is significant axonal loss. Inflammation is variable, sometimes absent. The pathology is most severe in proximal nerve segments and spinal roots and may not be full blown in the sural nerve biopsy. CIDP is thought to represent an autoimmune T-cell and antibody reaction against unknown myelin antigens. Its treatment consists of plasma exchange, intravenous immunoglobulin, and corticosteroids.
The GBS and CIDP are the counterparts of MS for the peripheral nervous system. They are important, because timely intervention with plasma exchange can prevent death in the GBS and severe permanent disability in CIDP. There are standardized criteria for their diagnosis, based on the clinical, CSF, nerve conduction, and biopsy findings.
HEREDITARY NEUROPATHIES
Seventy percent of neuropathies in children are inherited and 30% are acquired. The reverse is true of adults. The inherited neuropathies include lysosomal storage diseases, peroxisomal disorders, mitochondrial disorders, familial amyloidoses, and other entities. Neuropathy, in these diseases, is a component of a systemic metabolic defect. The inherited neuropathies include also two groups called Hereditary Motor and Sensory Neuropathy (HMSN) and Hereditary Sensory and Autonomic Neuropathy (HSAN), in which neuropathy is the main or only abnormality (see teble below).
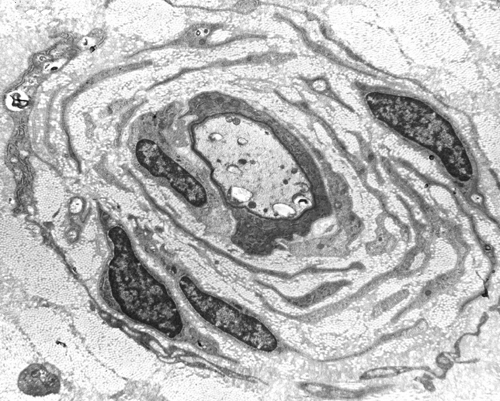 Hypertrophic neuropathy |
 Hypertrophic neuropathy |
The most common HMSN and the most common overall familial neuropathy is Charcot-Marie-Tooth disease (CMT). CMT affects 1 in 2500 persons and is autosomal dominant. It causes weakness and atrophy of distal muscles, especially those innervated by the peroneal nerve ("stork leg"), pes cavus, and sensory loss. It begins in childhood or adolescence and progresses slowly, involving other nerves. It is compatible with a normal lifespan. Nerve conduction studies show decreased conduction velocity. The nerve biopsy in CMT1 shows demyelination, myelin regeneration (thin myelin), axonal loss, and onion bulbs. In longstanding cases there is gross thickening of nerves, hence the term hypetrophic neuropathy.
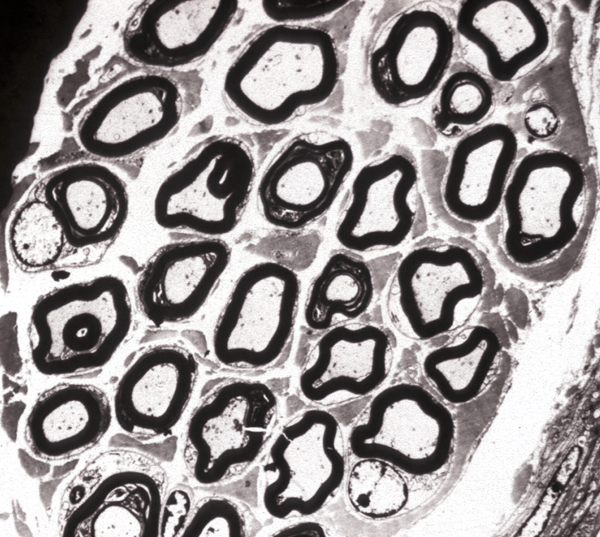 HSAN III |
The name CMT denotes a specific entity but is used also to encompass the entire group of HMSN. In that group, the phenotype described above is referred to as CMT1. Other, less frequent HMSNs are listed in the table. The HSANs are characterized by sensory dysfunction (depressed reflexes, altered pain and temperature perception) and variable autonomic dysfunction (GER, postural hypotention without tachycardia, excessive sweating). HSAN III (Familial Dysautonomia/Riley Day syndrome) in particular shows also hypothermia, no axon flare on intradermal histamine injection, lack of fungiform papillae in the tongue, and other abnormalities. Nerves in HSAN III lack small myelinated and unmyelinated axons, and sensory and autonomic ganglia show loss of ganglion cells.
INHERITED NEUROPATHIES
HSAN |
The HMSNs are characterized by great genetic heterogeneity. There are autosomal dominant, recessive, and X-linked forms. CMT1 alone is genetically diverse. Its most common autosomal dominant form is due to duplication of a segment of chromosome 17 (17p11.2-p12) that contains the gene for a 22 kd peripheral myelin protein, PMP22. This protein probably plays a role in Schwann cell differentiation. CMT1 patients have three copies of the normal gene and presumably produce 1.5 times as much PMP22 as normal people do. Other forms of CMT1 are recesssive or X-linked and are caused by point mutations of the PMP22, Cx32, and Myelin Protein Zero (MPZ) genes. Other HMSNs are equally diverse with mutations of the same gene causing different phenotypes and mutations of different genes causing the same phenotype.
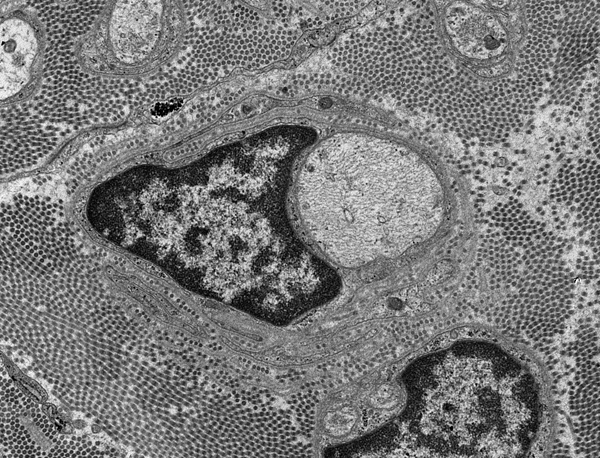 Congenital hypomyelinating neuropathy |
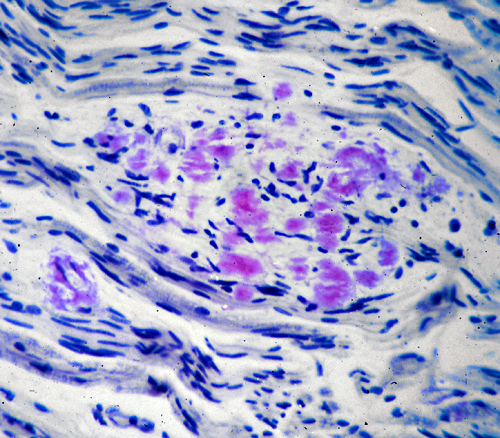 Amyloid neuropathy |
Familial amyloid neuropathies(FAP) are a group of familial systemic amyloidoses with involvement of peripheral nerves. The most common FAP is caused by an autosomal dominant mutation of the transthyretin gene on 18q11. The mutant protein is deposited in the form of amyloid and damages peripheral nerves, the heart, kidneys, gastrointestinal tract, and other organs. In nerves, amyloid damages first and most severely small fibers, causing loss of pain and temperature sensation and autonomic dysfunction. Transthyretin is produced in the liver. Liver transplantation arrests the progression of the disease.
Myeloma and other monoclonal plasma cell proliferations result in the production of abnormal amounts of immunoglobulins and immunoglobulin chains. In some of these conditions, immunoglobulins form amyloid deposits which damage nerve fibers mechanically. In others, immunoglobulins have antibody activity against MBP and other myelin proteins and cause a demyelinative neuropathy. Other plasma cell dyscrasias and dysproteinemias cause vasculitis.
VASCULITIC NEUROPATHY
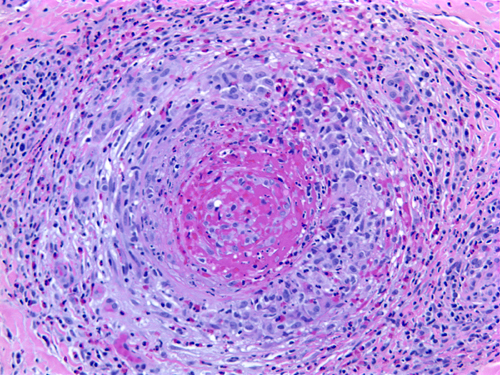 Necrotizing arteritis |
Polyarteritis nodosa and other vasculitides often involve peripheral nerves causing single or multiple mononeuropathies (due to nerve ischemia), asymmetric polyneuropathy, and distal symmetric polyneuropathy. A sural nerve biopsy along with a muscle biopsy are the best tissues for establishing the diagnosis of vasculitis. The nerve biopsy is diagnostic in over half of patients with systemic vasculitis and clinical neuropathy, and the diagnostic yield increases with the addition of a muscle biopsy. Such biopsies show necrotizing arteritis, perivascular inflammatory infiltrates, hemorrhage and hemosiderin deposition, neovascularization in epineurial arteries, and variable changes in nerve fascicles, depending on the severity and stage of neuropathy. The muscle shows vasculitis and denervation atrophy.
SMALL FIBER NEUROPATHY
Small fiber neuropathy (SFN) refers to a set of symptoms caused by damage of unmyelinated and small myelinated axons that convey pain and temperature sensation and serve autonomic functions. These symptoms are burning or shooting pain and tingling, loss of pain and temperature sensation, and autonomic abnormalities (loss of or excessive sweating, color and other skin changes) affecting the extremities in a stocking-glove distribution. There may also be visceral autonomic dysfunction (tachycardia, erectile dysfunction, constipation, urinary retention). The most common cause of SFN is diabetes and related entities, including impaired glucose tolerance and insulin-induced neuropathy. Other causes include vitamin deficiencies, drug toxicity, monoclonal gammopathy, hypothyroidism, alcoholism, Sjogren syndrome, Lyme disease, sarcoidosis, amyloidosis, Fabry disease, and paraneoplastic neuropathies. Some SFNs are genetic and are caused by sodium channel mutations and some are caused by autoantibodies to potassium channels. Many of these entities affect also larger nerve fibers and can cause weakness and loss of vibration and position sense. When large fibers are affected, electrodiagnostic studies are abnormal. If only small fibers are affected, which is often the case at the onset of these neuropathies, electrodiagnostic studies are normal, and the best way to confirm the diagnosis is a skin biopsy, stained with antibodies to Protein Gene Product 9.5, an axonal marker. Normal skin contains axons that cross the dermis, traverse the basement membrane, and branch into the epidemis. In SFN, these fibers are decreased or lost. Thus, epidermal nerve fiber density (ENFD) is a sensitive and reliable index of SFN.
SENSORY NEURONOPATHY (GANGLIONOPATHY)
Sensory neuronopathy (SN) is due to degeneration and loss of neurons in sensory ganglia and Wallerian degeneration of their distal axons (in peripheral nerves) and their proximal axons (in dorsal roots and the posterior columns of the spinal cord). Some SNs are associated with carcinoma, especially small cell carcinoma of the lung and other cancers, and some are seen in autoimmune disorders, such as Sjogren syndrome. Some are due to infection (HIV) or toxicity (pyridoxin, platinum chemotherapeutic agents). SN may be a component of inherited metabolic disorders, such as Friedreich ataxia and POLG deficiency. Most cases have no defined cause (idiopathic). The paraneoplastic SNs are thought to have an autoimmune basis supported by the finding of antineuronal (anti-Hu) autoantibodies in the serum and CSF. SN may precede the onset of the neoplasm and may be associated with encephalomyelitis. SNs have a subacute or chronic onset and cause sensory loss and ataxia as well as autonomic dysfunction (because of involvement of sympathetic ganglia). Unlike distal axonopathies which affect distal parts of the extremities first and more severely, SNs may affect any part of the body, including the face. Pathological examination of sensory ganglia shows lymphocytic inflammation and loss of ganglion cells, atrophy of dorsal roots, and degeneration of dorsal columns of the spinal cord. Nerve biopsy shows loss of myelinated axons without regenerating axon sprouts.
Further Reading
- Lauria G, Lombardi R. Skin biopsy: a new tool for diagnosing peripheral neuropathy. BMJ 2007;334:1159-62 PubMed
- Szigeti K, Lupski JR. Charcot-Marie-Tooth disease. Eur J Hum Genet 2009;17:703-10. PubMed
- Gabreels-Festen A:Dejerine-Sottas syndrome grown to maturity: overview of genetic and morphological heterogeneity and follow up of 25 patients. J Anat 2002;200:341-56.PubMed
- Gibbons CH. Small Fiber Neuropathies. Continuum (Minneap Minn) 2014;20:1398-1412 PubMed
- Axelrod FB, Gold-von Simson G. Hereditary sensory and autonomic neuropathies: types II, III, and IV. Orphanet J Rare Dis. 2007;2:39. PubMed
- Amato AA, and Ropper AH. Sensory ganglionopathy. New Engl J Med 2020;383:1657-62. PubMed
Updated: October, 2020