CEREBRAL CONTUSIONS, DIFFUSE AXONAL INJURY, AND THE SHAKEN BABY SYNDROME
CEREBRAL CONTUSIONS
 Contre- coup contusions |
A contusion is hemorrhagic necrosis of brain tissue. When the head is abruptly brought to a stop against a solid object, such as the dashboard or the ground, the brain continues to move for an instant, hitting the inside the now stationary skull. The soft brain is easily contused and lacerated by the hard bony ridges at the base of the skull or by the tentorium cerebelli and falx cerebri. Contusions usually involve the surface of the brain, especially the crowns of gyri, and are more frequent in the orbital surfaces of the frontal lobes and the tips of the temporal lobes. Acute contusions show hemorrhagic necrosis and brain swelling. Gradually, macrophages remove necrotic brain tissue and blood. Eventually, the contusion evolves into a yellowish plaque characterized by loss and atrophy of brain tissue, glial scarring, hemosiderin deposition, and loss of axons in the underlying white matter. A cerebral contusion can be distinguished from a cerebral infarct because, in the infarct, the superficial cortex is usually preserved, whereas in the contusion, it is the first to be destroyed. Contusions may correspond to the site of impact ("coup" contusions) -from the French word "blow"- or develop opposite the impact ("contre coup" contusions). In falls, contre coup are more frequent than coup contusions. A fall to the back with the occiput hitting the ground causes contusions in the inferior frontal and temporal lobes.
DIFFUSE AXONAL INJURY (TRAUMATIC AXONAL INJURY)
Diffuse axonal injury (DAI) is a special traumatic lesion, which occurs most frequently in motor vehicle accidents and following blows to the unsupported head. In the course of such injuries, the cerebrum goes into a back and forth gliding motion, pivoting around the upper brainstem. The brainstem, together with the cerebellum, is held firmly fixed by the tentorium, and the falx prevents side-to-side motion. Axons are stretched but do not snap from this injury; their sudden deformation causes changes in the axonal cytoskeleton (compaction of neurofilaments, fracture of microtubules) that lead to an arrest of the fast axoplasmic flow. Components of this flow, including mitochondria and other organelles, accumulate proximal to the lesion and cause axonal swellings (spheroids). Some axons with mild lesions probably recover but many eventually rupture. It takes several hours from trauma to axonal rupture. Influx of calcium through the stretched axolemma probably initiates the process that leads to the formation of spheroids. Mitochondrial dysfunction and neuroinflammation contribute to the local tissue injury. Ruptured axons undergo Wallerian degeneration leading to loss of neurological function. Loss of axons may lead to dying back of neurons. Thus, DAI is a multifaceted process that evolves over time. The swellings are located at nodes of Ranvier where the axolemma is more liable to deform because there is no myelin. Brain damage is most severe along midline structures (corpus callosum, brainstem) where the shear forces are greatest, and at the cortex-white matter junction because of the change in the consistency of brain tissue.
Clinically, patients with severe DAI become unconscious immediately after the injury and either remain comatose or go into a persistent vegetative state. In severe TBI, DAI is compounded by widespread vascular injury and other traumatic lesions which cause cerebral edema and HIE.
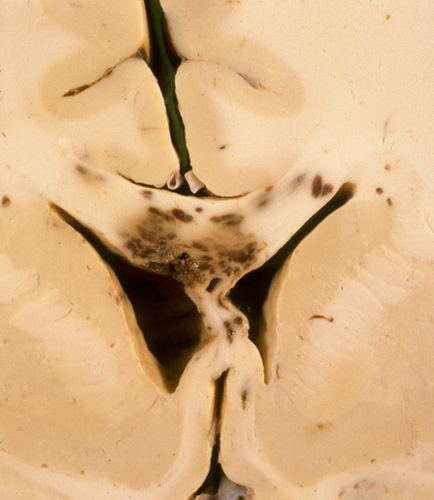 DAI: corpus callosum hemorrhages |
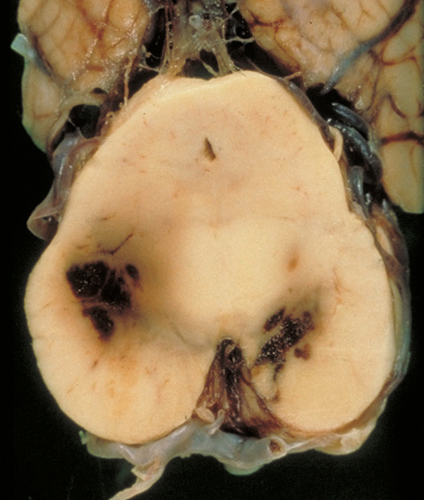 DAI: brainstem hemorrhages |
DAI is rarely a pure lesion, and, clinically, its effects are difficult to separate from other concurrent TBI pathology. When severe, it can depress consciousness in the acute phase and can cause lasting impairment of memory and cognition. The MRI shows small hypointense lesions corresponding to traumatic microbleeds. While being the only marker of DAI, the microbleeds may not correlate with the degree of axonal damage. In acute DAI, the brain is either normal or shows petechial hemorrhages in the corpus callosum, centrum semiovale, dorsolateral brainstem, and other areas, due to tearing of blood vessels. These vascular lesions should be distinguished from secondary brainstem hemorrhages that occur with herniations (see further on).
 DAI: axonal swellings. BAPP immunostain. |
 DAI: severe white matter degeneration. |
Microscopically, the damaged white matter shows axonal swellings. These can occur anywhere but are particularly common in the parasagittal parts of the brain, the corpus callosum, fornix, internal capsule, and the brain stem. Axonal swellings can be detected with H&E and silver stains 15 hours after the injury. Immunostains with antibodies to Beta Amyloid Precursor Protein (BAPP) can detect the axonal lesions in 2-3 hours after the injury. BAPP is produced by neurons as a reaction to injury. It flows down the axon and accumulates at points of axonal constriction or transection. Some axons have multiple swellings that give them a beaded appearance. Axonal swellings may persist for years. Distal to the swellings, axons and myelin degenerate and gliosis develops over time. Severe DAI may cause decrease of white matter volume, atrophy of the corpus callosum, and dilatation of the lateral ventricles. Some patients with severe TBI and DAI also have neuronal loss and microglial nodules in the cortex, deep nuclei and brainstem (diffuse neuronal injury.)
CEREBRAL CONCUSSION iis characterized by transient loss of consciousness and post-traumatic amnesia of variable duration, in most cases less than 30 minutes, and other symptoms, including anxiety, depression, sleep disorder and headache. Its pathology is difficult to establish because there is no opportunity for neuropathological observations. It had been considered to be a reversible functional disturbance without structural damage. It has been proposed that there is a continuum of brain injury in concussion, ranging from very mild cases in which patients are dazed for a few seconds without losing consciousness, to the most severe ones in which there are persistent neurological abnormalities and structural changes that blend with DAI. In that sense, classic cerebral concussion may be a mild form of DAI. Loss of neurological function is probably caused by axonal dysfunction. Loss of consciousness, in particular, may be caused by dysfunction of the reticular activating substance of the upper brainstem. This is the part of the CNS that is subjected to the highest twisting force during sagittal rotation of the hemispheres. More important, it has become evident that repeated concussions, especially before symptoms from the previous one resolve, have a cumulative effect and can cause chronic traumatic encephalopathy. While the focus has been on the white matter, MRI studies have shown recently reduced cortical thickness and reduced hippocampal volumes in children with mild concussion. These abnormalities persist up to 4 months postinjury and suggest structural damage to the gray matter.
The initial event in concussion is probably axonal distortion or stretching, which allows efflux of potassium into the extracellular space and inlfux of calcium into axons. This results in release of glutamate, which triggers an excitotoxic cascade, similar to hypoxic-ischemic encephalopathy. The Na+/K+ ATP-dependent pump overworks in order to restore ionic balance, leading to energy depletion, glycolysis, lactic acidosis, and oxidative dysfunction. There are severe fluctuations of cerebral perfusion. The effects of this neurometabolic cascade range from reversible neuronal depression to permanent damage.
CHRONIC TRAUMATIC ENCEPHALOPATHY
It has been known for many years that boxers develop a form of neurodegeneration that has been called the “punch-drunk” state and “dementia pugilistica”. In recent years, this condition has been reported in professional football players and renamed “Chronic Traumatic Encephalopathy” (CTE). In addition to boxers and football players, CTE has been reported in athletes involved in other contact sports and in other individuals with a history of repetitive head impacts. The syndrome of CTE begins insidiously, usually many years after the patients have stopped playing sports, with inattention, mood and behavior disturbances, confusion, and memory loss, and progresses inexorably over many years to a stage of full-blown dementia and parkinsonism. The brain, in CTE, shows atrophy, dilatation of the lateral and third ventricles, and thinning of the corpus callosum. Microscopic examination reveals hyperphosphorylated tau (p-tau) deposition primarily in neurons, around small vessels. These changes are patchy and affect the deeper parts of cerebral sulci. Astrocytes containing p-tau are also present in the subpial cortex but this finding is associated more with age and not with repetitive head impacts. Other neurodegenerative pathologies, including beta amyloid deposition in the form of diffuse or neuritic plaques, amyloid angiopathy, TDP-43-inclusions, and other may co-exist with p-tau deposition. It is clear that neuronal p-tau deposition is the key cellular change in CTE. The cause of CTE is thought to be TBI, especially repeated cerebral concussions and subconcussive trauma. In the acute phase, concussion, especially following side-to-side hits to the head, causes DAI and triggers the release of tau and beta amyloid in the brain. This, along with cerebral hypoxia, excitotoxicity and inflammatory mediators, set in motion a progressive destructive cascade that causes neurodegeneration many years later.
ABUSIVE HEAD TRAUMA IN INFANTS AND CHILDREN (THE SHAKEN BABY SYNDROME)
 Shaken baby syndrome: retinal and optic nerve hemorrhages. |
 Shaken baby syndrome: retinal and optic nerve hemorrhages. |
The term shaken baby syndrome (SBS) describes a pattern of child abuse which is the second most common cause of death in children under one year of age, after the sudden infant death syndrome. Grabbing an infant by the shoulders and shaking or jerking sets the head into a whiplash or dangling motion. The effects of this abuse are worse in infants because their heads are large and their neck muscles weak. Acceleration and deceleration of the brain causes severe traumatic brain injury and may also cause cervical spine injury. In severe cases, the mechanical effects are compounded by hypoxic and ischemic encephalopathy. While shaking alone is enough to cause the injury, many cases result from a combination of shaking and blunt impact or blunt impact alone, and the term "shaking-impact syndrome" has been used by some authors. The American Academy of Pediatrics has recommended the term abusive head trauma (AHT) which includes shaking and blunt impact. Thus, the SBS can be viewed as a subset of AHT. The SBS does not occur after trivial injury or rough handling. Substantial force is needed to produce brain damage. It can result from a single episode or repeated bouts of abuse. Infants with the SBS are usually brought to the Emergency Department with a nonspecific clinical picture of hypotonia, listlesness, vomiting, irritability, and lethargy, which may be mistaken for medical illness. Many are apneic and comatose or are dead on arrival. Survivors may suffer severe permanent brain damage. The diagnosis may be missed unless a skeletal survey and fundoscopic examination are done.
The SBS is characterized by a triad of encephalopathy, subdural hematomas and retinal hemorrhages. The subdural hematomas are located in the interhemispheric fissure or over the convexities and may be of varying ages. They are usually small initially but may enlarge later or additional subdurals may appear when brain atrophy develops in infants who survive.
Retinal hemorrhages, often large and confluent, are an important component of the SBS and correlate with the severity of brain damage. They are probably caused by shaking of the vitreous body which pulls the retina and tears its delicate vessels. Documentation of retinal hemorrhage by fundoscopic examination is important in the clinical evaluation of the SBS. Retinal hemorrhages are uncommon in accidental brain injury and are infrequently seen in patients with bleeding disorders, angiopathies, sepsis, and other nontraumatic settings.
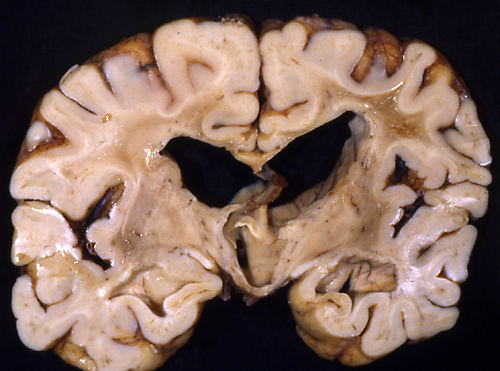
 SBS: severe brain atrophy secondary to HIE. |
 MRI of patient on left. |
The subdural hematomas and retinal hemorrhages are markers of the SBS. Encephalopathy and death are caused mainly by global HIE-the key pathology of the SBS-and increased intracranial pressure. The back and forth motion of the head during shaking applies great stress to the craniocervical junction and the lower brainstem. In extreme cases, the spinal cord may separate from the brainstem. Lesser forces impair the function of vital centers in the brainstem causing central apnea. Cerebral edema which follows HIE raises intracranial pressure and makes HIE worse. Vascular tears from shaking cause microscopic or large parenchymal hemorrhages. Infarcts in an irregular geographic pattern are common. These lesions add to the neurological deficits and further raise the intracranial pressure. The subdural hematomas may add another increment. DAI may affect the cortex-white matter junction, the corpus callosum, internal capsule, or the brainstem and contributes to the encephalopathy. Infants with the SBS usually also have rib, skull, and other fractures (often of varying ages), skin and soft tissue bruises, internal injuries, and other evidence of trauma.
INTRACEREBRAL HEMATOMA
Severe head trauma can also cause deep intracerebral hematomas and brain necrosis. Traumatic intracerebral hematomas are often multiple. They are found more commonly in the frontal and temporal white matter. They are probably due to rupture of intrinsic vessels as result of angular rotation of the brain.
POST-TRAUMATIC CEREBRAL ISCHEMIA
A large proportion of patients with severe or fatal TBI also have cerebral infarcts. Most of these infarcts are in vascular territories and a few affect watershed zones. The underlying causes are intracranial hypertension, vascular compression from herniations, vasospasm, traumatic vascular tears, dissecting hematomas and other vascular lesions. Global HIE is also a frequent finding in severe TBI. It is caused by a combination of systemic hypotension and intracranial hypertension, leading to cerebral hypoperfusion. Cardiovascular collapse and other systemic changes may result from the effects of DAI on the medulla. Infarcts and HIE greatly increase morbidity and mortality in TBI.
Further Reading
- Blumenthal I. Shaken baby syndrome. Postgrad Med J 2002;78:732–735. PubMed
- Marino R, Gasparotti R, Manzoni D, et al. Posttraumatic cerebral infarction in patients with moderate or severe head trauma. Neurology 2006;67:1165-71. PubMed
- Case ME. Inflicted Traumatic Brain Injury in Infants and Young Children. Brain Pathology 2008;18:571-82. PubMed
- McKee AC, Cantu RC, Nowwinski CJ, et al. Chronic Traumatic Encephalopathy in Athletes: Progressive Tauopathy After Repetitive Head Injury. J Neuropathol Exp Neurol 2009;68:709-35. PubMed
- Christian CW, Block R, and the Committee on Child Abuse and Neglect. Abusive Head Trauma in Infants and Children. Pediatrics 2009;123:1409–1411. PubMed
- Levin AV. Retinal Hemorrhage in Abusive Head Trauma. Pediatrics 2010;126;961-970. PubMed
- Barkhoudarian G, Hovda DA, Giza CC. The Molecular Pathophysiology of Concussive Brain Injury. Clin Sports Med 2011;30:33-48. PubMed
- McKee AC, Cairns NJ, Dickson DW, et al. The first NINDS/NIBIB consensus meeting to define neuropathological criteria for the diagnosis of chronic traumatic encephalopathy. Acta Neuropathol. 2016; 131: 75–86.PubMed
- Mez J, Daneshvar DH, Kiernan PT, et al. Clinicopathological Evaluation of Chronic Traumatic Encephalopathy in Players of American Football.JAMA 2017;318(4):360-370. PubMed
- Hill CS, Coleman MP, Menon DK. Traumatic Axonal Injury: Mechanisms and Translational Opportunities. Trends in Neurosciences 2016;39:311-24. PubMed
- Berkowitz CD. Physical Abuse of Children. N Engl J Med 2017;376:1659-66. PubMed
- Butler MLMD, Dixon E, Stein TD et al. Tau Pathology in Chronic Traumatic Encephalopathy is Primarily Neuronal. J Neuropathol Exp Neurol. 2022 Sep 19;81(10):773-780. doi: 10.1093/jnen/nlac065. PubMed
- Mayer A R, Meier T B, Dodd A B et al. Prospective Study of Gray Matter Atrophy Following Pediatric Mild Traumatic Brain Injury. Neurology 2023;100e516-e527
- McKee A C, Stein TD, Huber BR, et al. Chronic traumatic encephalopathy (CTE): criteria for neuropathological diagnosis and relationship to repetitive Impacts. Acta Neuropathologica (2023) 145:371–394 PubMed
Updated: February, 2023